Fig. 6.1
MPGN due to chronic infection (shunt nephritis). (a) Light microscopy showing an MPGN pattern of injury (PAS ×40). Immunofluorescence microscopy showing granular capillary wall staining for (b) IgG, (c) IgM, (d) C3, (e) kappa light chains, and (f) lambda light chains. Electron microscopy showing (g) capillary wall deposits and (h) subendothelial expansion with cellular elements and electron-dense deposits. Arrows point at deposits
Monoclonal Gammopathies (2000s)
Recently, it was shown that monoclonal gammopathies, with or without cryoglobulins, are another important cause of MPGN [33, 34]. Sethi et al. showed that 41 % of MPGN patients without an autoimmune process had serum and/or urine electrophoresis studies positive for a monoclonal gammopathy. Based on the bone marrow biopsy findings, the majority of these patients were classified as having a monoclonal gammopathy of undetermined significance. Other less common causes of MPGN due to deposition of monoclonal Ig included multiple myeloma, low-grade B cell lymphoma, and chronic lymphocytic leukemia. This was a retrospective study, and many of these patients were initially labeled as MPGN type I based on the MPGN pattern on LM, Ig deposits that were often monoclonal on IF, and subendothelial deposits with double-contour formation on EM. Many of these cases were not ascribed to an underlying monoclonal gammopathy even after the renal biopsy showed immunoglobulin restriction on IF [33]. Based on this study, it was suggested that in patients with MPGN and immunoglobulin restriction on IF, the monoclonal gammopathy should not be called of “undetermined significance” or MGUS, but rather these patients should be diagnosed as having a “monoclonal gammopathy related or monoclonal gammopathy associated MPGN” [33]. Other recent studies have also shown that monoclonal deposition of Ig, in particular IgG3, results in a proliferative glomerulonephritis [35]. It should be pointed out that in our experience deposition of monoclonal Ig other than IgG3 such as monoclonal IgA or IgM also result in MPGN [36]. Indeed, deposits composed of light chains alone in absence of any heavy-chain component can result in MPGN. Figure 6.2 shows an example of MPGN resulting from deposition of monoclonal immunoglobulins.
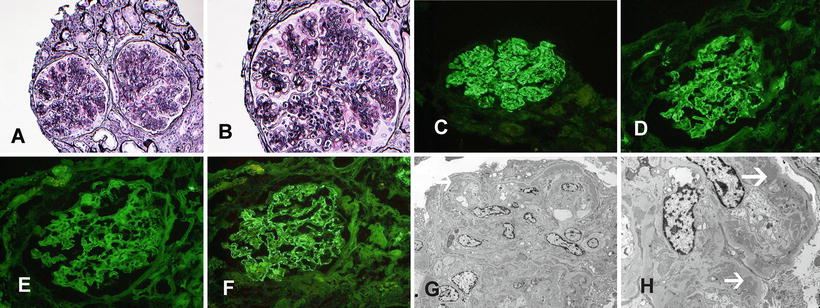
Fig. 6.2
MPGN due to monoclonal immunoglobulin deposition. MPGN due deposition of monoclonal immunoglobulins. (a, b) Light microscopy showing an MPGN pattern of injury (silver a ×20, b ×40). Immunofluorescence microscopy showing granular capillary wall staining for (c) IgG, (d) C3, and (f) kappa light chains. Note negative (e) lambda light chains. Electron microscopy showing (g, h) capillary wall deposits and subendothelial expansion with cellular elements and new basement formation resulting in double contours. Arrows point at deposits
Dysregulation of the Alternative Pathway of Complement (2000s)
Activation of the alternative pathway (AP) of complement occurs in a sequential manner that can be divided into four main steps: initiation of complement activation, C3 convertase activation and amplification, C5 convertase activation, and terminal pathway activity with assembly of the terminal complement complex or membrane attack complex (MAC). Once activated, the AP generates effector compounds that are delivered to all surfaces indiscriminately, mandating control over progression of the cascade and the action of these molecules. Multiple complement regulators and inhibitors operate at every level to prevent self-mediated damage. For example, proteins that regulate C3 convertase (C3bBb) assembly, activity, and half-life include factor H, factor I, factor B, decay-accelerating factor, factor H-related proteins, membrane cofactor protein (CD46), and complement receptor 1. Mutations in, or antibodies against, these proteins therefore can alter AP control and lead to the dysregulation of the AP.
Whatever the mechanism, dysregulation of the AP results in activated complement products that are delivered indiscriminately to all surfaces, including the glomerular capillary walls with subendothelial and mesangial deposition of the complement products and debris. For example, dysregulation of AP occurs when antibodies develop to proteins that regulate C3 convertase (C3bBb) assembly and activity such as factor H, factor I, factor B, decay-accelerating factor, factor H-related proteins such as factor H-related protein 5, membrane cofactor protein, and complement receptor 1 [37–41]. Similarly, mutations in these complement-regulating proteins, including C3 itself, can alter AP control leading to deposition of complement products along the glomerular capillary walls. Antibodies to C3 convertase (C3 nephritic factor or C3Nef) also prolong its half-life resulting in activation of AP of complement. Genetic background also is another risk factor for development of disease. Best studied are the Tyr402His allele variants of factor H. His402 (H402) is overrepresented in MPGN patients with alternative pathway abnormalities as compared to Tyr402, and functional studies have shown that the former provides poorer factor H-mediated regulation of the C3 convertase on cell surfaces (Table 6.1) [38–40, 42–54].
Table 6.1
Abnormalities of the AP of complement
Complement factor | Abnormality | Pathology | References |
|---|---|---|---|
C3 | C3 mutation | DDD | [42] |
Factor H | Factor H mutation | DDD and C3 glomerulonephritis | |
Factor I | Factor I mutation | C3 glomerulonephritis | |
Factor H | Antibody | DDD and C3 glomerulonephritis | |
Factor H-related protein 5 | CFHR5 mutation | C3 glomerulonephritis | |
Factor B | Antibody | DDD | [49] |
C3NeF | Antibody | DDD and C3 glomerulonephritis | |
Membrane cofactor protein/CD46 | Mutation | C3 glomerulonephritis | [38] |
Factor H alleles (p.Y402H, p.V62I) | Allele variant | DDD and C3 glomerulonephritis | |
C3 alleles (p.R102G, p.P314L) | Allele variants | DDD and C3 glomerulonephritis |
Once complement products and debris are deposited in mesangium and subendothelial region of the capillary walls, it leads to glomerular inflammation, manifested as a proliferative GN followed by reparative changes and development of MPGN.
The prototypical example of a glomerular disease associated with dysregulation of the AP is dense deposit disease (DDD) [50, 55, 56]. It is characterized by an MPGN pattern on LM, C3 deposition in the mesangium and along capillary walls on IF, and osmiophilic sausage-shaped wavy dense deposits along the GBM and in the mesangium on EM. The absence of significant Ig by immunofluorescence and the location and character of the dense EM deposits distinguish DDD from Ig-mediated MPGN. Laser microdissection of glomeruli from DDD patients followed by mass spectrometry of the glomerular proteins shows large amounts of complement factors of the AP and terminal pathway.
On the other hand, many cases labeled as MPGN type I or type III that show mesangial and capillary wall C3 staining (and no significant Ig deposits) with subendothelial (type I) and subepithelial deposits (type III) on EM also result from dysfunction of AP due to mutations or antibodies to the complement-regulating proteins [40]. These proliferative lesions are termed C3 glomerulonephritis or C3-GN [37, 57]. Figure 6.3 shows an example of MPGN resulting from complement deposition, in the absence of Ig deposits.
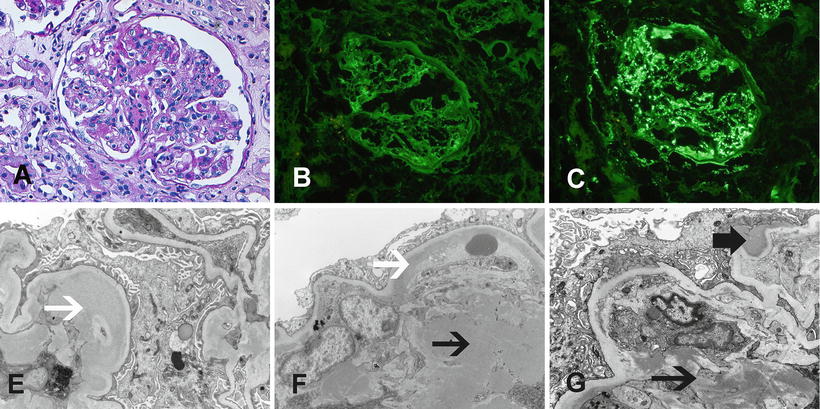
Fig. 6.3
MPGN due to C3 glomerulonephritis. MPGN due deposition of monoclonal immunoglobulins. (a) Light microscopy showing an MPGN pattern of injury (PAS ×40). Immunofluorescence microscopy showing (b) no staining for IgG, (c) bright mesangial and capillary wall staining for C3. Electron microscopy showing (d, e, f) numerous subendothelial, and mesangial deposits and occasional subepithelial deposits. Black arrows point at subendothelial deposits, white arrows point at mesangial deposits, and thick black arrow points at subepithelial deposit
Data from laser microdissection and mass spectrometry analysis of glomeruli obtained from a number of patients with MPGN type I and type III with C3 deposits, i.e., C3 glomerulonephritis, is consistent with unrestricted activation of the AP; the proteomic profile in these cases is similar to that found in patients with DDD [40, 54]. That DDD and C3-GN are part of a continuum is further supported by cases of MPGN with strong C3 deposition on IF that show features that are intermediate between DDD and C3-GN, with some capillary loops showing sausage-shaped intramembranous deposits, while other loops show subendothelial and subepithelial deposits on EM.
The limited data on C3-GN suggests that it carries a better prognosis than DDD [38, 40]. It has been hypothesized that increase in C3 convertase activity is associated with the DDD phenotype while increase in C5 convertase activity is associated with the C3-GN phenotype [58]. In addition, it is also likely that certain allele variations of complement-regulating proteins may be associated with DDD while others may be associated with C3-GN.
The differential diagnosis of MPGN due to AP dysfunction includes postinfectious glomerulonephritis (due to the presence of subepithelial humps) and autoimmune disease (due to subepithelial, subendothelial, and mesangial deposits). In fact, most of the cases of MPGN with C3 deposits that we receive in our practice have previously been labeled as either idiopathic MPGN type I/III or postinfectious glomerulonephritis (often healing or resolving). However, there is often no history of infection in these cases of MPGN that have been labeled as postinfectious GN. Subsequent evaluation of these cases in fact has shown that these patients have an MPGN due to dysfunction of the AP of complement. In these cases, the lack of significant Ig deposition on IF should alert the pathologist that they are dealing with complement-mediated MPGN instead of a postinfectious GN. Another helpful histological difference is that MPGN due to AP dysfunction shows large and abundant mesangial, subendothelial, and occasionally subepithelial deposits that have a homogenous amorphous lobular quality and typically few intramembranous deposits can be found (similar to DDD but not as dense), whereas capillary wall deposits in a postinfectious GN are often sparse, sharp, and well demarcated.
Finally, even though MPGN with C3 deposits due to AP dysfunction is being now recognized more frequently and is often separately classified as C3-GN, many cases of MPGN from AP dysfunction are still likely to be classified as MPGN type I and type III [37–40, 57, 59]. This is because based on the current classification, there is considerable overlap and some cases can belong to either group. Thus, it is important to understand that based on the current classification, MPGN with C3 deposits belonging to either MPGN type I/III or C3GN can all result from dysfunction of the AP of complement. Thus, this has necessitated the recent proposal of a new classification (next section).
Thrombotic Microangiopathy
Lastly, an MPGN pattern of injury can also result from damage to the endothelial cells in cases associated with thrombotic microangiopathies (TMA) [60]. In the acute phase, mesangiolysis, endothelial swelling, and fibrin thrombi are present in the glomerular capillaries. As the process evolves into a reparative and chronic phase, mesangial expansion and glomerular capillary wall remodeling with double-contour formation takes place, resulting in the classical double-contour pattern associated with late stages of MPGN. Thus, the healing phase of TTP/HUS, cases of aHUS associated with complement abnormalities [61], antiphospholipid antibody syndrome, drug-induced TMA, nephropathy associated with bone marrow transplantation, radiation nephritis, malignant hypertension, and connective tissue diseases can all present with an MPGN pattern of injury. In these cases, however, Ig and complement are typically absent on IF, and EM does not show electron-dense deposits along the capillary walls.
Proposal for a New Classification of MPGN
There is a recent proposal to classify MPGN based on the IF findings rather than the EM findings. Thus, MPGN has been classified as immune complex mediated and complement mediated [62]. Immune complex-mediated MPGN shows Ig and/or complement factors on IF studies. Complement-mediated MPGN shows complement factors and the lack of any significant Ig on IF studies. Immune complex-mediated MPGN includes MPGN resulting from chronic infections, autoimmune diseases, and dysproteinemias. On the other hand, complement-mediated MPGN results from dysregulation of the AP of complement. It should be noted that extensive complement (C3 and C4) deposition is also present in immune complex-mediated MPGN. However, the complement deposition in these cases results from activation of the classical and terminal pathway of complement by the immune complexes.
We recommend the terms MPGN types I–III should be abandoned since it implies a primary/idiopathic MPGN, while it is likely that an underlying etiology can be found in the majority of cases of MPGN.
Clinical Presentation
The clinical presentation of MPGN is variable. During the injury and proliferative phase, the presentation is often as nephritic syndrome. As the disease progresses and reparative changes set in, the presentation evolves to a nephrotic syndrome. In fact, many cases present with a nephritic-nephrotic syndrome. Thus, the phase of the disease process often dictates the clinical presentation. In addition, the different pathophysiological mechanisms that result in MPGN, i.e., immune complex versus complement deposition also likely contribute to the heterogeneity in the clinical presentation. This is further complicated by a variable histology, since immune complex and/or complement deposition results not only in an MPGN but can present as milder form with mesangial proliferative lesion or severe lesion with crescents [63, 64].
Thus, microscopic hematuria, mild proteinuria, or full-blown nephritic or nephrotic syndrome may occur depending on the type of the glomerular lesion as well as timing of the biopsy in relation to the clinical presentation. The same is true regarding the presence of hypertension and/or degree of kidney impairment.
Evaluation of MPGN
In view of the evidence discussed above, we propose that cases in which renal biopsy shows an MPGN pattern of injury, the work-up should be directed towards two major pathophysiologic factors: immune complex mediated and complement mediated. If Ig is present on IF, the evaluation should include evaluation for infections, autoimmune diseases, and monoclonal gammopathies, including cryoglobulins. On the other hand, if IF shows predominantly C3 and is negative or shows no significant staining for Ig, then the MPGN should be worked up as a complement-mediated MPGN, which can be further subdivided into DDD or C3-GN based on the results of the electron microscopy examination. Complement-mediated MPGN warrants an in-depth study of the AP, regardless of whether the renal biopsy shows DDD or C3-GN. We predict that Ig-mediated MPGN is more likely to be present in adults whereas complement-mediated MPGN is more likely to be present in children and young adults. It is likely that a genetic mutation in the complement regulatory proteins of the AP accounts for the many cases of a complement-mediated MPGN noted in children and young adults, while cases occurring in adults are more likely to be secondary to the development of autoantibodies to one of the complement-regulating proteins. Furthermore, in few cases of complement-mediated MPGN, an infection, autoimmune disease, or a monoclonal gammopathy may result in the development of antibodies or monoclonal proteins that are directed against epitopes on complement-regulating proteins, thus leading to dysfunction of the AP of complement. However, in such cases, the MPGN will be due to AP dysfunction not due to Ig deposition, and the renal biopsy will show C3 without any significant Ig deposition [51]. On the other hand, few cases of immune complex-mediated MPGN may also have dysfunction of the AP of complement. Finally, it is possible that even after extensive evaluation of the two groups, we will be unable to determine the underlying etiology in a small number of immune complex-mediated MPGN or complement-mediated MPGN [65].
We believe that new tests/techniques will continue to reveal new causes for a lesion that until recently was considered to be idiopathic in a significant number of patients. Table 6.2 shows the proposed evaluation of the AP of complement.
Table 6.2
Proposed evaluation of MPGN secondary to AP dysfunction
Screening tests | C3 and C4 levels AH50 and CH50 sMAC levels AP functional and hemolytic assay |
Mutations/genetic analyses, including allele variants | C3 Factor H Factor I Factor B MCP/CD46
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




