foreign surfaces such as some microbial products and damaged cells. AP activation begins at C3 without involving C1, C4, or C2. In individual complement-mediated diseases, several of these pathways may be involved.5,6 Among the immunoglobulins, IgG subclasses IgG1 and IgG3 and IgM are classical C pathway activators, whereas IgG2 and IgG4 and normally glycosylated IgA activate C poorly.7 However, C activation and its sequelae need not involve immunoglobulin deposits and may occur by other mechanisms even in the presence of immunoglobulin deposits. All C activation pathways lead to cleavage of C5 and release of chemotactic factors such as C5a that attract inflammatory cells (neutrophils, macrophages, platelets) when activation occurs within, or adjacent to, the circulatory compartment. Cleavage of C5 by C5 convertases also leads to the release of C5b and the addition of C6, C7, C8, and multiple C9 molecules to form the lipophilic terminal membrane attack complex (MAC or C5b-9) (Fig. 45.1).8,9 Sublytic quantities of C5b-9 can insert into lipid bilayers of adjacent glomerular cell membranes and act in a fashion similar to receptor agonists. Sublytic C5b-9 initiates several signaling pathways and thus converts endothelial cells, mesangial cells, and podocytes to local inflammatory effector cells that can proliferate; release a variety of cytokines, growth factors, eicosanoids, oxidants, proteases, and other acute inflammatory mediators; as well as upregulate genes that encode matrix components and contribute to chronic overproduction of the extracellular matrix with scarring and sclerosis.9 Immunoglobulin-induced C activation products like C5a can also activate TLRs, thus linking the innate and adaptive immune systems (Fig. 45.2).10 C activation in vivo is tightly regulated by a number of circulating and cell-bound C regulatory proteins (CRPs) the functions of which, particularly those of CR1, factor H, membrane cofactor protein (MCP), and CD59, are also important in the development of several glomerular diseases (Fig. 45.2).5,6
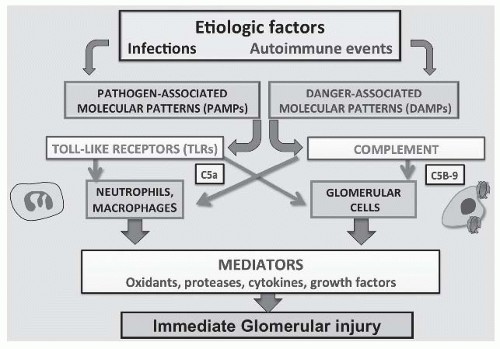 FIGURE 45.1 The innate immune system in glomerular disease. Etiologic factors, usually infections or autoimmune events, are presented to the immune system as pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs). These interact with the two principal arms of the innate immune system, Toll-like receptors, and the complement system. Toll-like receptors are present on both circulating inflammatory cells like neutrophils and macrophages and on resident glomerular cells including endothelial and mesangial cells and podocytes. Toll-like receptor activation induces inflammation through release of multiple mediators including cytokines, growth factors, oxidants, proteases, eicosanoids, and others. Activation of complement also leads to the attraction of inflammatory cells through the generation of chemotactic factors such as C5a and to the conversion of resident glomerular cells to inflammatory cells following sublytic C5b-9 insertion into cell membranes. The mediators released by both infiltrating neutrophils and macrophages, and resident glomerular cells cause glomerular injury that leads to morphologic and functional changes in diseased glomeruli. |
there is an initial phase of antigen excess leading to the formation of small, soluble immune complexes followed by equivalence with midrange size complexes that are believed to be trapped in tissues, and finally, to antibody excess with large complexes that are cleared through the mononuclear phagocyte system with eventual elimination of the antigen. Glomerular deposits of antigens and antibodies in acute serum sickness form primarily in the mesangium and in a subendothelial distribution. In chronic serum sickness, repeated antigen administration is provided to maintain slight antigen excess and is associated with more subepithelial deposits, again attributed to the passive glomerular trapping of small, preformed immune complexes that crossed the capillary wall to localize beneath podocytes.12,13 Attribution of the tissue injury that occurs in serum sickness to the passive trapping of circulating complexes followed from observations that inflammation occurred only during the period that complexes could be detected in the circulation, corresponded with appearance of immune deposits in glomeruli and that both the antigen and antibody components of the immune complexes were constituents of the deposits.13 However, later studies using passively administered preformed immune complexes did induce some glomerular deposits, but generally failed to replicate the tissue injury seen in either acute or chronic serum sickness.14 Moreover, later measurements of circulating immune complex levels in human disease found little correlation between circulating complex levels, sizes, and disease activity. Other studies done in antiglomerular antibody (nephrotoxic nephritis [NTN]) models rather than in serum sickness demonstrated that antibody deposits activated C and other mediators that attracted circulating inflammatory cells—primarily neutrophils—which then caused tissue injury.15 Some of these discrepancies were resolved in 1978 when it was shown that the classic subepithelial immune complex deposits in the Heymann models of membranous nephropathy (MN) in rats formed in situ and were unrelated to circulating immune complexes. Instead, they resulted from antibody binding to endogenous glomerular components localized on the podocyte (see Membranous Nephropathy, which follows).14,16,17,18 Subsequent studies in both acute and chronic serum sickness using cationic BSA confirmed that deposits in these models involving exogenous antigens also formed locally and were unrelated to circulating immune complex levels or sizes.19,20 This new paradigm allowed the mediation of immune complex nephritis to be studied directly rather than by being extrapolated from findings in models of anti-GBM disease.
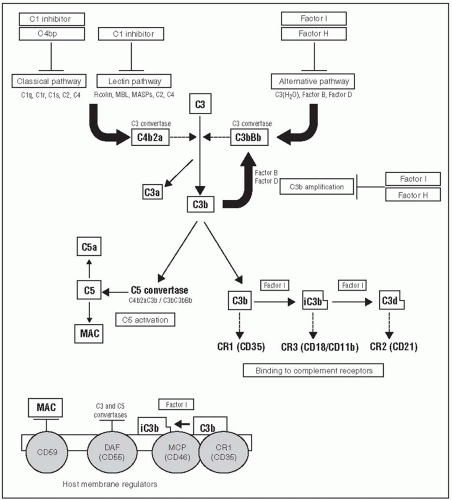 FIGURE 45.2 A schematic depiction of complement pathways and how they are activated as they relate to the pathogenesis of glomerulonephritis (GN). Activation via the classical, lectin, and alternative pathways leads to the formation of C3 convertases that cleave C3 to C3a and C3b. C3b can interact with complement receptors on cell surfaces such as CR1, CR2, and CR3 as well as contribute to the formation of C5 convertase. Cleavage of C5 results in the formation of the chemotactic factor C5a and C5b, which leads to the formation of the C5b-9 membrane attack complex (C5b-9) that is important in the mediation of several glomerular diseases. Factors H and I are circulating regulators of the alternative complement pathway, whereas CD59, decay accelerating factor (DAF), membrane cofactor protein (MCP), and complementary regulatory 1 (CR1) serve as membrane-bound regulators that protect cells from complement attack. MBL, mannose-binding lectin; MASP, MBL-associated serum protease; MAC, membrane attack complex (C5b-9) (Reproduced with permission from Pickering M, Cook HT. Complement and glomerular disease: new insights. Curr Opin Nephrol Hypertens. 2011;20(3):271-277.) |
“one shot” serum sickness model in rabbits, leading to a prolonged search for the “nephritogenic” streptococcal antigen that has extended over several decades. Although many candidate proteins have been proposed, most have failed to meet strict criteria for causality such as being demonstrable in deposits, particularly subepithelial “humps,” inducing antibody that correlated with clinical disease, and inducing a similar disease in animal models.32 However, streptococcal pyogenic exotoxin B (SpeB) meets most of these criteria. SpeB is a small (28 kDa), cationic (pK 9.3) cysteine protease with C activating and plasmin-binding properties and represents 90% of the secreted extracellular protein made in vivo by nephritogenic strains of group A streptococci.32 Antibody to SpeB correlates with disease activity in PSGN and colocalizes with IgG and C3 in subepithelial “humps.”32,33 However, the intense exudative glomerular inflammatory response and subepithelial humps are not well explained by the analogy to acute serum sickness because intact circulating ICs do not form subepithelial IC deposits directly, and subepithelial IC deposits do not produce inflammation, presumably because complement activation products like C5a go directly into the urine and are not chemotactic for cells in the circulation.14,18 Moreover, IgG is sometimes absent, or only a minor constituent of the deposits, whereas C3 deposition has been reported to both precede and exceed detectable IgG.34,35 Several explanations for these apparent contradictions are plausible. They include observations that some subendothelial deposits are also present by electron microscopy (EM) in PSGN,34,35 perhaps because the antibody to SpeB exhibits molecular mimicry with endothelial cell antigens, and antiendothelial antibody deposits are generally rapidly cleared.36 In addition, SpeB alone is a zymogen that can activate C directly through the MBL pathway independent of IgG.32 SpeB also exhibits plasmin-binding properties that can facilitate C activation and might cause proteolysis of GBM and facilitate the transit of dissociated subendothelial ICs to form subepithelial humps.37 Finally, PSGN often exhibits autoimmune features including both IgM and IgG rheumatoid factors with cryoglobulin activity, antiendothelial antibodies, anti-DNA antibodies, and antineutrophil cytoplasmic antibodies (ANCA). Although the respective roles of these nonstreptococcal antibodies in mediating the disease, if any, remain undefined,
these findings suggest an autoimmune component to postinfectious GN that is consistent with current thinking about most other immune glomerular diseases (Table 45.1).38,39,40
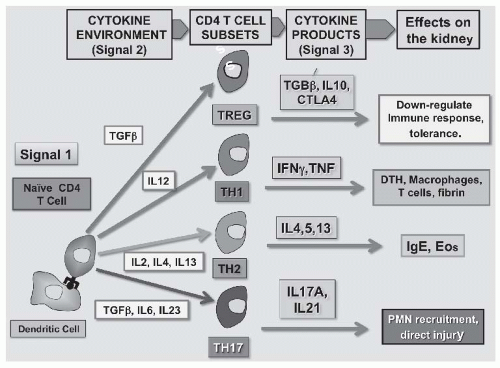 FIGURE 45.3 The T-cell component of the adaptive immune system in glomerulonephritis (GN). Antigen is presented to naïve CD4 T cells by dendritic cells (Signal 1). Depending on the predominant cytokine environment, T cells differentiate into CD4 T-cell subsets that play different roles in the pathogenesis of glomerular disease. In the presence of transforming growth factor beta (TGF-β), T regulatory cells (Tregs) develop that make TGF-β, interleukin 10 (IL-10), and cytotoxic T-lymphocyte antigen 4 (CTLA-4) that downregulate and control the immune response. IL-12 stimulates differentiation into Th1 cells that make interferon gamma (IFNγ) and TNF and produce traditional T-cell/macrophage-mediated delayed-type hypersensitivity (DTH) reactions. IL-2, IL-4, and IL-13 favor the development of Th2 cells that make IL-4, IL-5, and IL-13, and lead to allergic-type hypersensitivity reactions involving immunoglobulin E (IgE) and eosinophils (Eos). The CD4 T cells most implicated in the pathogenesis of GN are Th17 cells that differentiate in the presence of TGFβ, IL-6, and especially IL-17, and those that produce IL-17a and IL-21 that facilitate recruitment of other inflammatory cells such as neutrophils (PMNs) and also cause tissue injury directly. |
TABLE 45.1 The Most Common Complement Profiles and Autoimmune Features in Glomerulonephritis | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
the bloodstream, it might also reach the circulation if abnormal trafficking of these cells to the bone marrow occurs.49 Underglycosylated IgA1 undergoes conformational change and exhibits altered biologic properties compared to normal IgA1, including increased tendencies to self-aggregate, to activate C, and to bind to other molecules like fibronectin, IgG, and collagen IV.45,46,50 In circulating macromolecular form, the underglycosylated IgA1 aggregates lack the glycosylated sites necessary to interact with asialoglycoprotein and the CD 89 receptors in the liver and spleen, thus evading normal clearing mechanisms and facilitating mesangial localization.45,46,51 It is not yet known if the “lanthanic” mesangial IgA deposits seen in 6% to 16% of normal donor kidneys without disease contain underglycosylated or normal IgA.42
extrarenal events have any role in autoimmunization is not known.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


