Fig. 3.1
Tears are able to enter the lacrimal sac but not to exit because of a one-way valve effect. Pressure in the lacrimal sac increases with dilatation of the sac and formation of an intranasal cyst if an imperforated membrane at the valve of Hasner is present
Infection is common, occurring in 24–60 % of dacryoceles [18, 20]. The acute dacryocystitis associated with dacryocystoceles is different from the low-grade dacryocystitis associated with typical NLDO [19] because it begins earlier in life (within the first 1–2 weeks) and is more severe. If untreated, patients may develop an abscess within the lacrimal sac, and preseptal cellulitis may occur that could lead to orbital cellulitis, sepsis, and meningitis. Therefore, more aggressive systemic antibiotic treatment is indicated for neonates with infections.
Dacryocele has a high rate of spontaneous resolution, with 50 % of dacryoceles imaged prenatally resolving prior to birth [21]. Medical treatment of dacryoceles with massage of the tear sac and topical antibiotics may result in resolution in up to 76 % of dacryoceles [18]: in patient without improvement or spontaneous resolution or in case of infection, the best treatment is lacrimal probing with excision of the intranasal cyst. Probing alone has success rate from 50 to 100 % because the intranasal cyst, just opened without removal, could close again.
The presence of an enlarged erythematous mass overlying the lacrimal sac in a newborn is usually adequate to establish the diagnosis of an infected dacryocystocele. If the dacryocystocele is not infected or inflamed, the typical location supports the diagnosis. However, other possible diagnoses need to be considered. Dacryocystoceles present beneath the medial canthus; they may extend onto the medial lower eyelid, but they do not arise directly on the eyelid. Other features that help establish the diagnosis include the absence of globe displacement, the absence of nasal deformities, and the presentation of dacryocystoceles at birth or within the first 1–2 weeks after birth [19]. Lesions included in the differential diagnosis are encephalocele which usually presents above the medial canthus, with/without bulbar displacement; tumors arising from the orbit, brain, facial bones, nose, and paranasal sinuses; cutaneous hemangioma that is typically noted after 1–2 weeks from birth and has a vascular appearance either bright red or purplish, with the presence of hemangiomas elsewhere in the body and without symptoms of NLDO; dermoid and epidermoid cysts without symptoms of NLDO; and orbital lymphangiomas that cause proptosis.
Imaging of infants with dacryocystoceles is usually not necessary. However, it may be useful in patients with atypical features in whom the diagnosis is in question [19] or in cases without improvement after repeated treatments. The most useful are computed tomography with/without dacryocystography, ultrasounds, and magnetic resonance.
3.2.3 Congenital Lacrimal Puncta and Lacrimal Canaliculi Diseases
Congenital lacrimal diseases involving the lacrimal punctum or the lacrimal canaliculus consist of different diseases varying from agenesis to simple membrane occlusion that display clinically with epiphora. In proximal lacrimal system dysgenesis, single punctal/canalicular rather than bicanalicular (i.e., upper and lower) involvement was more common [2].
Agenesis of one or more lacrimal puncta is a rare finding, and it could be associated with the absence of the underlying canaliculus as reported by Lyons et al. [22]. This condition could be part of a systemic syndrome as it occurs in ectodermal dysplasia, Hay–Wells and Levy–Hollister syndrome.
The previously named as “punctal membranes,” now incomplete punctal canalization (IPC) as introduced by Ali et al. [23], is one of the most frequent and mild proximal lacrimal tract dysgenesis. The patient displays epiphora and it could be noticed as a translucent membrane. The authors [23] showed two typical locations of the membranes. The external membrane variety (IPC-EM) typically covers the external surface of the punctum and hides it beneath, giving a false impression of punctal agenesis. The internal membrane variety (IPC-IM) typically demonstrates blurred punctal margins but, just at the entry into the punctum, covers it entirely with a membrane. The authors did not find any systemic associations with IPC, although associated lacrimal system anomalies like canalicular stenosis and CNLDO were noted.
The pathogenesis of punctal membranes is not clear but is believed to either represent failed dehiscence of the epithelium overlying the normally formed canaliculi or a failure of canalization of the most proximal part of the nasolacrimal apparatus [2, 22].
Management modalities that have been elucidated in the literature include a punctoplasty with membrane lysis and intubation or retrograde marsupialization during DCR. Membranotomy using a simple punctum dilator is almost always helpful, generally without needing of intubation since the diameter of the punctum is fairly large following the procedure and does not tend toward restenosis later on [23]. Intubation is helpful if punctal membrane is associated with canalicular stenosis.
Supernumerary puncta and canaliculi are a rare congenital anomaly, and their incidence is not known [24]. In the majority of publications, they have been described as incidental examination findings in asymptomatic individuals. The effect that an additional lower punctum and canaliculus has on tear drainage is not known: in adults, functional impairment of canalicular drainage and acquired nasolacrimal duct obstruction may be contributory.
Supernumerary puncta and canaliculi are thought to arise from incomplete separation of the core from the surface epithelium or from abnormal out-budding of the proximal portion of the buried epithelial cord. Congenital double puncta may be associated with a predisposition to developing acquired nasolacrimal duct obstruction [24]. Unilaterality, predilection for the lower lid and medial siting of the accessory punctum are most recurrent descriptive findings [24].
3.3 Acquired Diseases
3.3.1 Nasolacrimal Duct and Lacrimal Sac Diseases
In the majority of cases, the etiology of an acquired obstruction in the lacrimal drainage apparatus remains obscure. Infection is a common associate, especially with obstructions involving the nasolacrimal duct. It is still not clear whether the infection is causal or sequential to the obstruction, or whether these two factors are simply part of a wider diathesis [25].
In particular, distal drainage system obstructions are idiopathic in 80 % of cases, and they are referred to as “PANDO” (primary acquired nasolacrimal duct obstruction) as a vascular aspecific inflammation, more frequent in female, that determines venous alterations with progressive atrophy or fibrosis of the lacrimal sac.
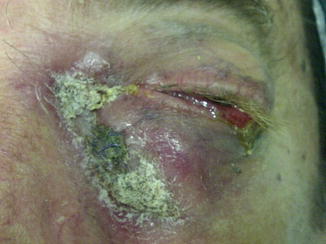
Obstructions of the upper portion of the nasolacrimal duct, where the lacrimal sac goes through the bony canal, are generally related to aspecific inflammations of the lacrimal sac (dacryocystitis). Dacryocystitis could be classified as acute, subacute, or chronic. This process could be limited to the lacrimal sac or involve the adjacent tissues and evolve to a pericystitis or an orbital cellulitis. The beginning of the process is characterized by vascular congestion, lymphoid cells infiltration, and edema. When localized to the lacrimal sac, a painful palpable swelling is clear at the medial canthus (Fig. 3.2); if an infection occurs, being S. aureus the most involved pathogen, the lacrimal sac could expand laterally, determining an obstruction of the nasolacrimal duct and of the lacrimal canaliculi, making the sac unreducible and causing retention of static fluid. This retention could become chronic with exacerbations, and it could lead to the formation of concretions and dacryoliths [26].
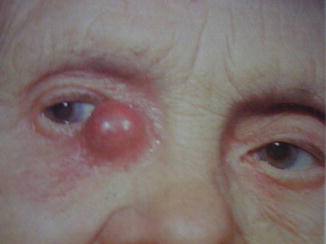
Fig. 3.2
Acute dacryocystitis
In dacryocystitis with pericystitis, the infection spreads through the sac to the surrounding tissues causing palpebral edema. If the infection goes beyond the orbital septum, an orbital cellulitis occurs with exophthalmos, bulbar dislocation, motility impairment, possible optical neuropathy, and loss of vision.
Obstructions of the lower portion of the nasolacrimal duct are generally subordinate to inflammatory processes from the inferior nasal meatus.
The etiology of the obstruction could vary from involutive changes, to recurrent inflammatory or infective processes (viral as herpes simplex virus, bacterial as Staphylococcus, fungal) rising from the nose and paranasal sinuses or from the eye, traumatic events or surgical procedures (Fig. 3.3), topic administrations of antiglaucoma and antiviral drops [27], radiotherapy, lacrimal sac or lacrimal duct tumors or neoplasms deriving from adjacent structures (orbit, nose, and paranasal sinuses), presence of foreign bodies (lashes, silicon), intranasal cocaine abuse [28], and systemic administrations of drugs that may have a toxic effect on the epithelium as fluorouracil or docetaxel [29].
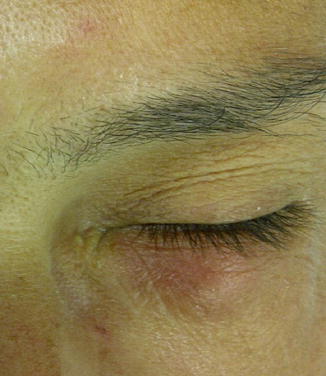
Fig. 3.3
Left lower lid inflammation incidental to contrast liquid wrong injection
Specific obstructions are those found in inflammatory diseases as Rosai–Dorfman syndrome [30], sarcoidosis [31–33], or Wegener’s granulomatosis [34]. Wegener’s granulomatosis is a multisystem inflammatory disease of unknown etiology. Ophthalmic involvement includes conjunctivitis, episcleritis, scleritis, corneal ulceration, uveitis, retinal vasculitis, and optic neuropathy; nasolacrimal duct obstruction has been reported in 7 % of patients with this disease [34] probably as a direct extension of nasal disease. In sarcoidosis, a systemic chronic granulomatous disease of unknown etiology with the involvement of the sinonasal mucosa and lacrimal drainage system is relatively uncommon and may run an independent course from ocular or systemic manifestations [31, 33]. Both these pathologies present a high rate of failure of DCR if it is not associated with the positioning of a silicon stent and a long-term therapy with local and systemic corticosteroids [32]. Primary lacrimal system tumors are rare but most of these (75–90 %) are malignant [35]. They encountered squamocellular carcinoma (the most common), transitional cell carcinoma, inverted papilloma, pleomorphic adenoma, oncocytoma and oncocytic carcinoma, adenocystic carcinoma, adenocarcinoma, mucoepidermoid carcinoma, malignant melanoma, sarcoma, and lymphoid cell tumors (Fig. 3.4).