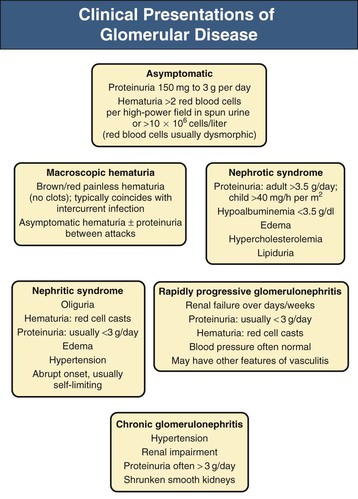
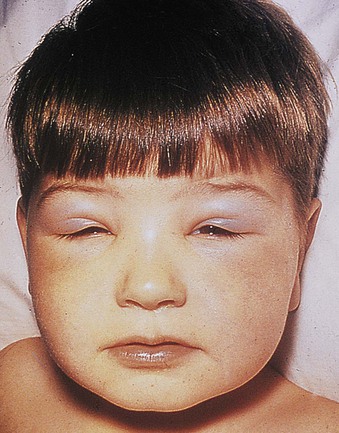
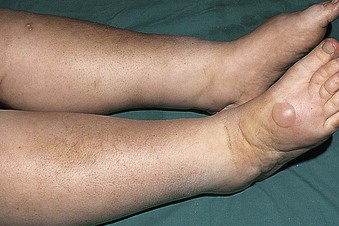
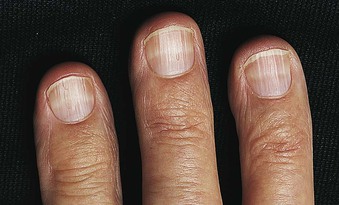
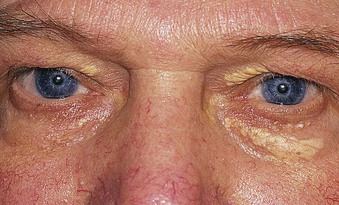
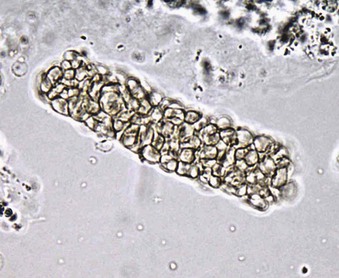
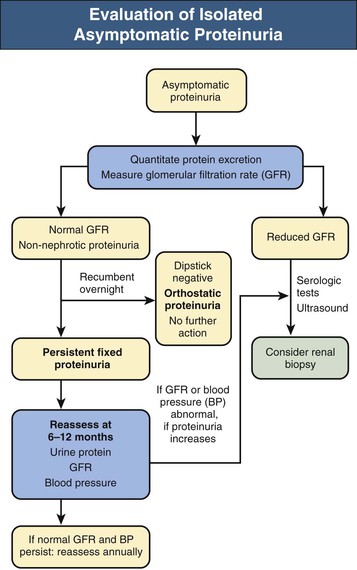
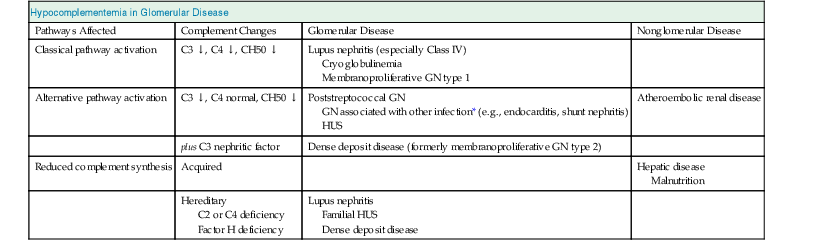
Jürgen Floege, John Feehally Glomerular disease has clinical presentations that vary from the asymptomatic individual who is found to have hypertension, edema, hematuria, or proteinuria at a routine medical assessment to a patient who has fulminant illness with acute kidney injury possibly associated with life-threatening extrarenal disease (Fig. 15-1). The most dramatic symptomatic presentations are uncommon. Asymptomatic urine abnormalities are much more common, but less specific, and may also indicate a wide range of nonglomerular urinary tract disease. The history, physical examination, and investigations are aimed at excluding nonglomerular disease, finding evidence of associated multisystem disease, and establishing renal function. The majority of glomerular diseases do not lead to symptoms that patients will report. However, specific questioning may reveal edema, hypertension, foamy urine, or urinary abnormalities during prior routine testing (e.g., during scheduled medical examinations). Multisystem diseases associated with glomerular disease include diabetes, hypertension, amyloid, lupus, and vasculitis. Apart from the individual history suggestive of these diseases, a positive family history may also be obtained in some cases. Other causes of familial renal disease may include Alport syndrome, especially if associated with hearing loss (see Chapter 48); uncommon familial forms of IgA nephropathy (see Chapter 23); focal segmental glomerulosclerosis (FSGS; see Chapters 18 and 19); complement-mediated glomerulonephritis (see Chapter 22); hemolytic uremic syndrome (HUS; see Chapter 29); and other rare conditions (see Chapter 28). Morbid obesity can be associated with FSGS. Certain drugs and toxins may cause glomerular disease, including nonsteroidal anti-inflammatory agents (NSAIDs) and interferon in minimal change disease (MCD); penicillamine, NSAIDs, and mercury (e.g., in skin-lightening creams) in membranous nephropathy; pamidronate and heroin in FSGS; and cyclosporine, tacrolimus, mitomycin C, and oral contraceptives in HUS. Recent or persistent infection, especially streptococcal infection, infective endocarditis, and certain viral infections (see Chapters 21, 57, and 58), may also be associated with a variety of glomerular diseases. Malignant neoplasms associated with glomerular disease include lung, breast, and gastrointestinal (GI) carcinoma in membranous nephropathy; Hodgkin disease in MCD; non-Hodgkin lymphoma in membranoproliferative glomerulonephritis; and renal carcinoma in amyloid disease (see Chapter 27). Patients will occasionally present with the renal disease as the first manifestation of a tumor. The presence of dependent pitting edema suggests the nephrotic syndrome, heart failure, or cirrhosis. In the nephrotic patient, edema is often periorbital in the morning (Fig. 15-2), whereas the face is not affected overnight in edema associated with heart failure (edema distributes by gravity, and patient with heart failure cannot lie flat because of orthopnea resulting from pulmonary congestion) or cirrhosis (patient cannot lie flat because of pressure on the diaphragm from ascites). As it progresses, edema of genitals and abdominal wall becomes apparent, and accumulation of fluid in body spaces leads to ascites and pleural effusions. Edema is unpleasant, leading to feelings of tightness in the limbs and a bloated abdomen, with practical problems of clothes and shoes no longer fitting. Surprisingly, however, edema may become massive in nephrotic syndrome before patients seek medical help; fluid gains of 20 kg (44 lb) or more are not unusual (Fig. 15-3). The edema becomes firm and stops pitting only when it is longstanding. In children, fluid retention may also be striking with nephritic syndrome. Chronic hypoalbuminemia is also associated with loss of normal pink color under the nails, resulting in white nails or white bands if the nephrotic syndrome is transient (Muehrcke lines, Fig. 15-4). Xanthelasmas may also be present as a result of the hyperlipidemia associated with longstanding nephrotic syndrome (Fig. 15-5). The presence of pulmonary signs should suggest one of the pulmonary-renal syndromes (see Boxes 24-3 and 24-4). Palpable purpura may be seen in vasculitis, systemic lupus, cryoglobulinemia, or endocarditis. Assessment of renal function and careful examination of the urine are critical (see Chapters 3 and 4). The quantity of urine protein and the presence or absence of dysmorphic red cells and casts will help classify the clinical presentation (see Fig. 15-1). Certain serologic tests are also helpful. These include antinuclear and anti-DNA antibodies for lupus, cryoglobulins and rheumatoid factor suggesting cryoglobulinemia, anti–glomerular basement membrane (anti-GBM) antibodies for Goodpasture disease, antineutrophil cytoplasmic autoantibody (ANCA) for vasculitis, and antistreptolysin O titer or streptozyme test for poststreptococcal glomerulonephritis. Serum and urine electrophoresis will detect monoclonal light chains or heavy chains and assays for free light chains in serum or urine may aid in their quantification, as in myeloma-associated amyloid or light-chain deposition disease. Testing for the presence of ongoing bacterial or viral infections is also useful. This includes blood cultures and testing for hepatitis B, hepatitis C, and human immunodeficiency virus (HIV) infection. Measurement of systemic complement pathway activation by testing for serum C3, C4, and CH50 (50% hemolyzing dose of complement) is often helpful in limiting the differential diagnosis (Table 15-1). Table 15-1 Hypocomplementemia in glomerular disease. * Glomerulonephritis (GN) with visceral abscesses is generally associated with normal or increased complement (elevations occur because complement components are acute-phase reactants). CH50, 50% Hemolyzing dose of complement; HUS, hemolytic uremic syndrome. The emerging role for genetic evaluation in patients with glomerulonephritis (GN) is discussed in Chapters 19 and 22. Ultrasound scanning is recommended in the workup to ensure the presence of two kidneys, to rule out obstruction or anatomic abnormalities, and to assess kidney size. Renal size is often normal in GN, although large kidneys (>14 cm) are sometimes seen in nephrotic syndrome associated with diabetes, amyloid disease, or HIV infection. Large kidneys can also occasionally be seen with any acute severe GN. The occurrence of small kidneys (<9 cm) suggests advanced chronic kidney disease (CKD) and should limit enthusiasm for renal biopsy or aggressive immunosuppressive therapies. Renal biopsy is generally required to establish the type of glomerular disease and to guide treatment decisions (see Chapter 6). In some patients, however, renal biopsy is not performed. If nephrotic children have no unusual clinical features, the probability of MCD is so high that corticosteroids can be initiated without biopsy (see Chapter 17). In patients with acute nephritic syndrome, if all features point to poststreptococcal GN, especially in an epidemic, biopsy can be reserved for the minority who do not show early spontaneous improvement (see Chapter 57). In Goodpasture disease (see Chapter 24), the presence of lung hemorrhage and rapidly progressive renal failure with urinary red cell casts and high levels of circulating anti-GBM antibody establishes the diagnosis without the need for a biopsy, although a biopsy may still provide valuable prognostic information. In patients with systemic features of vasculitis, a positive ANCA titer, negative blood cultures, and a tissue biopsy specimen from another site showing vasculitis are sufficient to secure a diagnosis of renal vasculitis. Again, however, renal biopsy may provide important clues to disease activity and chronicity. Biopsy is also not generally performed in patients with longstanding diabetes with characteristic findings suggestive of diabetic nephropathy and other evidence of microvascular complications of diabetes (see Chapter 30). Biopsy may also not be indicated in many patients with glomerular disease presenting with minor, asymptomatic urine abnormalities and well-preserved renal function because the prognosis is excellent, and histologic findings will not alter management. Urine testing that detects proteinuria or microhematuria is often the first evidence of glomerular disease. The random nature of urine testing in most communities inevitably means that much mild glomerular disease remains undetected. In some countries, symptomless individuals may have a urine test only if they require medical approval for some key life event, such as obtaining life insurance, joining the armed forces, or sometimes for employment purposes. In other countries, for example, Japan, urinalysis is performed routinely in school or for employment. These different practices may partly account for the apparently variable incidence of certain diseases, such as IgA nephropathy. Asymptomatic low-grade proteinuria and microhematuria and the combination of the two, increase in prevalence with age1 (Fig. 15-6). Nevertheless, there is no evidence to justify routine population-wide screening for asymptomatic urine abnormalities as renal biopsy and therapeutic intervention are rarely required when renal function is preserved. Screening, in particular for microalbuminuria, may be indicated for high-risk populations, for example, patients with diabetes, hypertension, or cardiovascular disease, and those with a family history of renal disease. Microhematuria is defined as the presence of more than two red blood cells (RBCs) per high-power field in a spun urine sediment (3000 rpm for 5 minutes) or more than 10 × 106 RBCs/l. Microhematuria is common in many glomerular diseases, especially IgA nephropathy and thin basement membrane nephropathy, although there are many other causes of hematuria (see Chapter 48). A glomerular origin should especially be considered if more than 5% of RBCs are acanthocytes (see Chapter 4) or if the hematuria is accompanied by RBC casts or proteinuria (Fig. 15-7). Glomerular hematuria is thought to result from small breaks in the glomerular basement membrane that allow extravasation of RBCs into the urinary space. This may occur in the peripheral capillary wall but more often occurs in the paramesangial basement membrane, particularly in diseases involving injury to the mesangium (mesangiolysis). As long as the renal tubules are intact, low amounts of serum proteins lost together with RBCs in damaged glomeruli can be fully reabsorbed, resulting in “isolated” microhematuria. The evaluation of microhematuria, discussed further in Chapters 48 and 61, begins with a thorough history. Urine culture should exclude urinary or prostatic infection. Phase contrast microscopy should follow in cases of persistent microhematuria to search for dysmorphic RBCs and RBC casts. In the absence of urine infection or a bladder catheter, any detectable proteinuria in the patient with asymptomatic microhematuria virtually excludes “urologic” bleeding and strongly suggests a glomerular origin. If this evaluation is nondiagnostic, renal imaging is performed to exclude anatomic lesions such as stones, tumors, polycystic kidneys, or arteriovenous malformations. In those older than 40 years who have persistent isolated microhematuria without evidence of a glomerular origin (see previous discussion), cystoscopy is mandatory to exclude uroepithelial malignant disease. In people younger than 40 years, such malignant disease is so rare that cystoscopy is not recommended. If all the prior study results are normal, a glomerular etiology is likely.2 The glomerular etiology can be determined only by renal biopsy, but this is rarely done because the prognosis is excellent in patients with normal renal function, normal blood pressure, and low-grade proteinuria (<0.5 g/day). However, repeated evaluation and prolonged follow-up are mandatory. The hallmark of glomerular disease is the excretion of protein in the urine. Normal urine protein excretion is less than 150 mg/24 h, consisting of 20 to 30 mg of albumin, 10 to 20 mg of low-molecular-weight proteins that undergo glomerular filtration, and 40 to 60 mg of secreted proteins (e.g., Tamm-Horsfall, IgA). Proteinuria is identified and quantified by dipstick testing or by assay in timed urine collections; Chapter 4 discusses test interpretation. Microalbuminuria is defined as the excretion of 30 to 300 mg of albumin per day, equivalent to a urine albumin to creatinine (g/g) ratio of 0.03 to 0.3, and is detected by quantitative immunoassay or by special urine dipsticks because this is below the sensitivity of the normal dipstick (see Chapter 30). This measurement is primarily used to identify diabetic subjects at risk for development of nephropathy and to assess cardiovascular risk, for example, in patients with hypertension. Non-nephrotic proteinuria is usually defined as a urine protein excretion of less than 3.5 g/24 h or a urine protein-creatinine ratio of less than 3 g/g. Whereas nephrotic-range proteinuria is absolutely characteristic of glomerular disease, asymptomatic proteinuria (<3.5 g/24 h) is much less specific and may occur with a wide range of nonglomerular parenchymal diseases as well as with nonparenchymal renal and urinary tract conditions that must be excluded by clinical evaluation and investigation. Increased urine protein excretion may result from alterations in glomerular permeability or tubulointerstitial disease, although only in glomerular disease will it be in the nephrotic range. Protein excretion can also increase from increased filtration through normal glomeruli (overflow proteinuria). Overflow proteinuria is typical of urinary light-chain excretion. It is seen in myeloma but can occur in other settings (e.g., release of lysozyme by leukemic cells) and should be suspected when the urine dipstick is negative for albumin despite detection of large amounts of proteinuria by other tests. Tubulointerstitial disease can be associated with low-grade proteinuria (usually <2 g/day). In addition to the loss of tubular proteins such as α1– or β2-microglobulin, there will also be some albuminuria due to impaired tubular reabsorption of filtered albumin. Tubular proteinuria accompanying glomerular proteinuria is an adverse prognostic sign in various glomerular diseases because it usually indicates advanced tubulointerstitial damage. Glomerular proteinuria is further classified into transient or hemodynamic (functional), that present only during the day (orthostatic), and persistent or fixed. Functional proteinuria refers to the transient non-nephrotic proteinuria that can occur with fever, exercise, heart failure, and hyperadrenergic or hyperreninemic states. Functional proteinuria is benign, usually assumed to be hemodynamic in origin and the result of increases in single-nephron flow or pressure. In children and young adults, low-grade glomerular proteinuria may be orthostatic, meaning that proteinuria is absent when urine is generated in the recumbent position. If there is no proteinuria in early-morning urine, the diagnosis of orthostatic proteinuria can be made. In patients with fixed orthostatic proteinuria, renal plasma flow and glomerular filtration rate (GFR) decrease in the upright position because of a lower systemic blood pressure. As recently proposed, the decreased GFR translates into a lower streaming potential across the filtration barrier, and albumin can no longer be excluded as efficiently from the filter by electrophoresis.3 Total urine protein in the patient with orthostatic proteinuria is usually less than 1 g/24 h; hematuria and hypertension are absent. Renal biopsy usually shows normal morphology or occasionally mild glomerular change. The prognosis is uniformly good, and renal biopsy is not indicated.4 Fixed non-nephrotic proteinuria is usually caused by glomerular disease. If GFR is preserved and proteinuria is less than 0.5 to 1 g/day, biopsy is not indicated but prolonged follow-up is necessary if significant proteinuria persists, to rule out the possibility of disease progression. Previous studies indicate that the biopsy findings in these patients can be similar to those seen in nephrotic syndrome (e.g., FSGS or membranous GN), although milder lesions are more common, particularly mesangial proliferative GN or IgA nephropathy. In general, other than regular monitoring and blood pressure control as needed, no treatment is necessary. Although it is controversial, many nephrologists will perform a renal biopsy in patients with normal GFR if non-nephrotic proteinuria exceeds 1 g/day, in particular if it persists after initiation of angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB) therapy. Figure 15-8 summarizes the evaluation of isolated asymptomatic proteinuria. Patients with coincident asymptomatic hematuria and proteinuria have a much greater risk of significant glomerular injury, hypertension, and progressive renal dysfunction. Minor histologic changes are less common. Renal biopsy is indicated even if urine protein is only 0.5 to 1 g/24 h if there is also persistent microhematuria with casts. Episodic painless macrohematuria associated with glomerular disease is often brown or “smoky” rather than red, and clots are unusual. Macrohematuria must be distinguished from other causes of red or brown urine, including hemoglobinuria, myoglobinuria, porphyrias, consumption of food dyes (particularly beetroot), and intake of drugs (especially rifampin). Macrohematuria caused by glomerular disease is observed primarily in children and young adults and rarely is seen after age 40. Most cases are caused by IgA nephropathy, but hematuria may occur with other glomerular and nonglomerular renal diseases, including acute interstitial nephritis. Although macrohematuria is typically painless, the patient may have an accompanying dull loin ache that suggests other diagnoses, such as stone disease or loin-pain hematuria syndrome. In IgA nephropathy, the frank hematuria is usually episodic, occurring within a day of an upper respiratory infection. The clear distinction between this history and the 2- to 3-week latency between an upper respiratory tract infection and hematuria is highly suggestive of postinfectious (usually poststreptococcal) GN; furthermore, patients with poststreptococcal disease usually will have other features of nephritic syndrome. Macrohematuria requires urologic evaluation, including cystoscopy, at any age unless the history (as shown previously) is characteristic of glomerular hematuria. Nephrotic syndrome is pathognomonic of glomerular disease. It is a clinical syndrome with a characteristic pentad5 (see Fig. 15-1). Patients may be nephrotic with preserved renal function, but in many, progressive renal failure will become superimposed when nephrotic syndrome is prolonged. Independent of the risk of progressive renal failure, the nephrotic syndrome has far-reaching metabolic effects that can influence the general health of the patient. Fortunately, some episodes of nephrotic syndrome are self-limited, and a few patients respond completely to specific treatment (e.g., corticosteroids in MCD). For most patients, however, it is a relapsing or chronic condition. Not all patients with proteinuria above 3.5 g/24 h will have a full nephrotic syndrome; some have a normal serum albumin concentration and no edema. This difference presumably reflects the varied response of protein metabolism; some patients sustain an increase in albumin synthesis in response to heavy proteinuria that may even normalize serum albumin. Table 15-2 shows the major causes of nephrotic syndrome. Proteinuria in the nephrotic range in the absence of edema and hypoalbuminemia has similar etiologies. The relative frequency of the different glomerular diseases varies with age (Table 15-3). Although it is predominant in childhood, MCD remains common at all ages.6 Prevalence of FSGS in African Americans has increased, which may explain why FSGS is becoming more common in U.S. adults but not in European adults.7,8 Table 15-2 Common glomerular diseases presenting as nephrotic syndrome in adults. HIV, Human immunodeficiency virus; NSAIDs, nonsteroidal anti-inflammatory drugs; PLA2R, phospholipase A2 receptor.
Introduction to Glomerular Disease
Clinical Presentations
Definition
Clinical Evaluation of Glomerular Disease
History
Physical Examination
Laboratory Studies
Hypocomplementemia in Glomerular Disease
Pathways Affected
Complement Changes
Glomerular Disease
Nonglomerular Disease
Classical pathway activation
C3 ↓, C4 ↓, CH50 ↓
Lupus nephritis (especially Class IV)
Cryoglobulinemia
Membranoproliferative GN type 1
Alternative pathway activation
C3 ↓, C4 normal, CH50 ↓
Poststreptococcal GN
GN associated with other infection* (e.g., endocarditis, shunt nephritis)
HUS
Atheroembolic renal disease
plus C3 nephritic factor
Dense deposit disease (formerly membranoproliferative GN type 2)
Reduced complement synthesis
Acquired
Hepatic disease
Malnutrition
Hereditary
C2 or C4 deficiency
Factor H deficiency
Lupus nephritis
Familial HUS
Dense deposit disease
Imaging
Renal Biopsy
Asymptomatic Urine Abnormalities
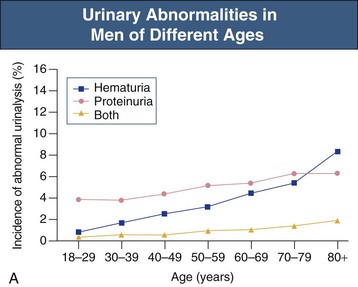
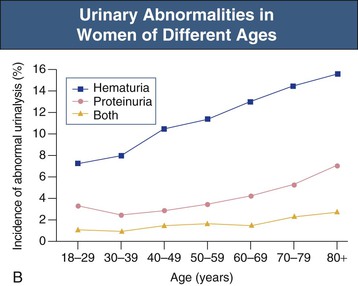
Asymptomatic Microhematuria
Pathogenesis
Evaluation
Asymptomatic Non-nephrotic Proteinuria
Overflow Proteinuria
Tubular Proteinuria
Glomerular Proteinuria
Functional Proteinuria
Orthostatic Proteinuria
Fixed Non-nephrotic Proteinuria
Asymptomatic Proteinuria with Hematuria
Macrohematuria
Nephrotic Syndrome
Definition
Etiology
Common Glomerular Diseases Presenting as Nephrotic Syndrome in Adults
Disease
Associations
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

Introduction to Glomerular Disease
Chapter 15