Gentzon Hall, Michelle P. Winn
Inherited Causes of Nephrotic Syndrome
Inherited nephrotic syndromes are rare diseases that present with nephrotic syndrome and varying degrees of renal impairment, but at times may also present with subnephrotic proteinuria. Inherited disease may manifest in utero or shortly after birth, as in congenital nephrotic syndromes, or later in life with proteinuria and pathologic findings consistent with focal segmental glomerulosclerosis. FSGS as the cause of nephrotic syndrome in adults is increasing according to North American (but not European) biopsy studies, and some believe that up to 18% of FSGS cases are caused by hereditary disorders.1 Autosomal dominant and recessive renal diseases result from defects in podocytes, the slit diaphragm, and the glomerular basement membrane (GBM). These primary renal disorders by definition do not recur after kidney transplantation. In children, syndromic conditions may be seen in which mutations in transcription factors involved in the development of multiple organ systems also affect renal morphogenesis and result in glomerulopathy. Further insight into these hereditary diseases has been elucidated through animal models, in which mutations in corresponding proteins often result in FSGS and proteinuria. Understanding these mechanisms not only provides better insight into abnormalities involved in idiopathic proteinuric kidney diseases but also allows the possible development of molecular drug targets that may improve the course of the renal disease and delay progression.2–4
Autosomal Recessive Diseases
Congenital Nephrotic Syndrome of the Finnish Type
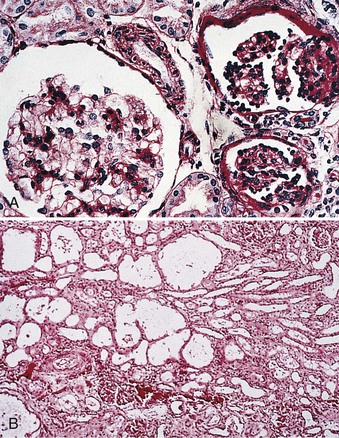
In 1956, Hallman described a disorder of massive proteinuria in utero in the Finnish population. Congenital nephrotic syndrome of the Finnish type (CNF) or nephrotic syndrome type 1 (NPHS1) is an autosomal recessive disease. It has an incidence of 1 in 8200 births in Finland and has been reported less frequently in other ethnicities. The placenta is larger than the child at birth, and the baby is usually born prematurely. Affected children will have as much as 30 g of proteinuria daily, massive edema, hypoalbuminemia, and hyperlipidemia. On light microscopy, kidneys typically have cystic dilation of the proximal tubules and diffuse mesangial sclerosis (Fig. 19-1). Effacement of podocyte foot processes and loss of the glomerular slit diaphragm are seen with electron microscopy.2
The product of the mutated NPHS1 gene, nephrin, is specifically expressed in podocytes, localized to the slit diaphragm between podocyte foot processes.3 Nephrin is a transmembrane protein belonging to the immunoglobulin superfamily with an intracellular, transmembrane, and extracellular domain. The extracellular domain forms the zipper-like structure of the slit diaphragm; the short intracellular domain interacts with the podocyte proteins podocin and CD2-associated protein (CD2AP). The most common mutations in nephrin are termed Fin-major and Fin-minor and account for 95% of the disease, but more than 50 other mutations in nephrin have been reported.4 NPHS1 knockout mice have massive proteinuria, have no slit diaphragm, and die within 24 hours of birth.5
Patients with CNF do not respond to cytotoxic therapy or corticosteroids. Angiotensin-converting enzyme (ACE) inhibitors and nonsteroidal anti-inflammatory drugs (NSAIDs) have no effect on reducing proteinuria in those with the Fin-major mutation, but there are some reports of efficacy of these agents in those with other mutations in nephrin. The ultimate goal of treatment of CNF is renal transplantation because without this approach, mortality is almost 100%. Because of overwhelming urinary loss of protein, affected infants without the Fin-major mutation may require medical nephrectomy with ACE inhibitors and NSAIDs, followed by renal replacement therapy before transplantation.
Corticosteroid-Resistant Nephrotic Syndrome
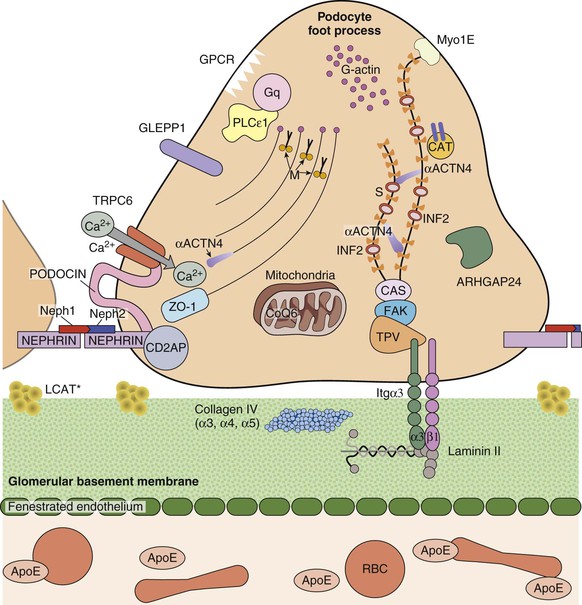
In children with nephrotic syndrome of unknown etiology, 90% respond to corticosteroids. Of the remaining 10%, many have corticosteroid-resistant nephrotic syndrome. Multiple hereditary forms have been identified. Mutations of NPHS2, which encodes the essential slit diaphragm protein podocin, cause an autosomal recessive disease seen mostly in children younger than 5 years.6 Affected children have nephrotic-range proteinuria, extrarenal complications of massive proteinuria (see Chapter 15), and rapid progression to end-stage renal disease (ESRD). On light microscopy, the renal lesions vary along the spectrum of minimal change disease to FSGS.4 Podocin is a hairpin-shaped transmembrane protein specifically expressed in podocytes2 (Fig. 19-2). Podocin localizes to the foot processes and seems to be involved in podocyte structure as well as in intracellular signaling, with the recruitment of nephrin and CD2AP to microdomains along the slit diaphragm.4
Corticosteroid-resistant nephrotic syndrome attributed to the NPHS2 mutation affects children between 3 months and 5 years of age. The age when the disease manifests seems to depend on the specific podocin mutation. The presence of at least one podocin mutation that codes for a stop codon or in patients homozygous for the R138Q mutation results in renal disease at about age 2 years. Two missense podocin mutations cause disease just before 5 years. Furthermore, studies demonstrate podocin mutations in some adult-onset FSGS. These patients must have two podocin mutations, one of which must be the podocin R229Q mutation.7
Another cause of corticosteroid-resistant nephrotic syndrome is a mutation in the phosphatase receptor type O (PTPRO), also called glomerular epithelial protein 1 (GLEPP1).8 PTPRO is a 150-kD transmembrane protein expressed on the apical membrane surface of podocytes. PTPRO deficiency has been demonstrated to alter podocyte structure and cause foot process flattening. Homozygous donor splice-site mutations result in skipping of PTPRO exon 16 or 19. Patients with PTPRO mutations present in the first decade of life with resistant nephrotic syndrome. In immunohistochemical analyses, the exon 16 mutation did not produce a change in the staining pattern of PTPRO in biopsy specimens, whereas the PTPRO staining with the exon 19 deletion biopsy specimens was completely absent, consistent with a lack of protein expression secondary to degradation of the messenger RNA product. Ultrastructural analyses of biopsy specimens revealed podocyte foot process effacement and microvillous transformation consistent with findings in the PTPRO−/− mouse model. The specific effects of the PTPRO mutations in podocytes are unknown.8,9
Isolated Diffuse Mesangial Sclerosis
Diffuse mesangial sclerosis is a pathologic finding in some patients with early-onset nephrotic syndrome. It occurs either “syndromic,” when it is associated with a particular clinical syndrome, or “isolated.” The isolated disease presents as nephrotic syndrome in the first month of life with rapid progression to ESRD. Light microscopy shows podocyte hypertrophy, mesangial matrix expansion, thickened basement membranes, and decreased size of glomerular capillary lumina. Some cases have shown response to cyclosporine.10 Diffuse mesangial sclerosis is inherited in an autosomal recessive pattern (Table 19-1). The causative gene PLCE1 encodes phospholipase C epsilon 1 (PLCε1). PLCε1 is a phospholipase involved in the generation of diacylglycerol and inositol 1,4,5-trisphosphate, which are intracellular second messengers (see Fig. 19-2). A PLCE1 knockout model in zebrafish revealed edema and pathologic characteristics of nephrotic syndrome during development. The mechanism of disease from the PLCE1 mutation is not clear, but evidence shows that it interacts with a guanosine triphosphatase (GTPase)–activating protein involved in podocyte development and interaction with nephrin. PLCE1 is not the only gene responsible for isolated diffuse mesangial sclerosis. More genetic causes are under investigation.10
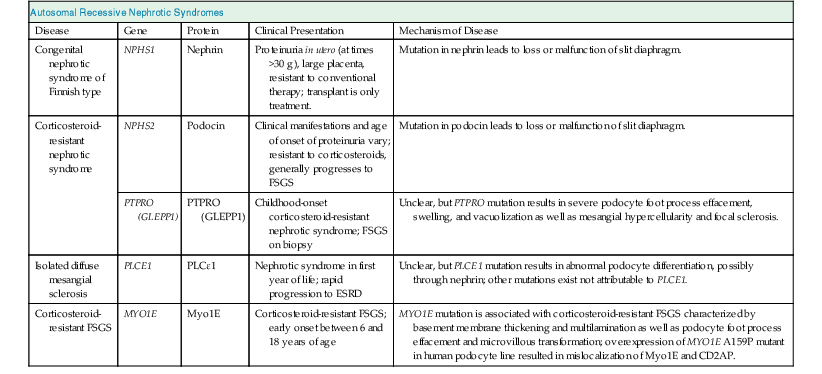
Autosomal Recessive Familial Focal Segmental Glomerulosclerosis
In 2011, mutations of MYO1E, which encodes the Myo1E protein, a nonmuscle class I myosin with important functions in the regulation of podocyte cytoskeletal dynamics, were identified as causes of nonsyndromic autosomal recessive FSGS.11 Mutations of MYO1E resulted in early-onset ESRD secondary to FSGS. These mutations have been associated with presentation between 1 and 9 years of age. Clinical signs and symptoms include nephrotic-range proteinuria and microhematuria, as well as hypoalbuminemia and edema. Renal biopsy findings are consistent with FSGS. Treatment with corticosteroids, ACE inhibitors, and cyclosporine A can result in partial remission, although some patients progress to ESRD. In vitro studies showed overexpression in a conditionally immortalized podocyte cell line that resulted in mislocalization of Myo1E and its interaction partner CD2AP. Also, targeted MYO1E knockout in mice produced basement membrane thickening and podocyte foot process effacement. Myo1E protein is postulated to be important in the regulation of tension development in podocyte foot processes, which may affect their ability to counterbalance variations in the glomerular capillary intraluminal pressure and maintenance of slit diaphragm integrity.11
Autosomal Dominant Diseases
Autosomal Dominant Familial Focal Segmental Glomerulosclerosis
In contrast to inherited nephrotic syndromes that present in childhood, there are familial proteinuric kidney diseases that manifest in adolescence through adulthood. Most of these are inherited in an autosomal dominant manner (Table 19-2). Typical pathology reveals the glomerular changes of FSGS, and there is variability in the rate of progression of renal impairment. Genetic mutations have been identified, and mechanisms of FSGS in sporadic forms can be elucidated from increasing understanding of these familial cases.12 Mutations of ACTN4 cause an autosomal dominant form of FSGS. The product of this gene is α-actinin 4, which is expressed in podocytes and cross-links with F-actin filaments in the foot processes (see Fig. 19-2). The mutation in ACTN4 is believed to be a gain-of-function mutation, leading to increased cross-linking of F-actin with α-actinin 4, resulting in dysregulation of actin assembly and disassembly in the podocyte. It was recently found that the relaxation frequency from actin of mutant ACTN4
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


