Immunoglobulin A Nephropathy
- Immunoglobulin A nephropathy (IgAN) is the most frequent form of primary glomerulonephritis worldwide.
- Hematuria is typical. Younger patients often present with episodes of macroscopic hematuria coincident with mucosal infections or exercise. Adults often have asymptomatic hematuria and proteinuria.
- Abnormal glycosylation of immunoglobulin A (IgA) molecules is believed to be important in disease pathogenesis.
- The diagnosis is established by the presence of dominant or codominant IgA deposition by immunofluorescence microscopy typically in the mesangial region of the glomeruli.
- True IgA nephropathy is not just “benign recurrent hematuria.” End-stage renal disease (ESRD) occurs in 15% of patients by 10 years and 20–40% by 20 years from onset.
IgAN is the most common form of primary glomerulonephritis worldwide. The prevalence of the disease shows geographic variations. In patients who undergo renal biopsy, IgAN accounts for approximately 30–40% of cases in Asia, 15–20% of cases in Europe, and 5–10% of cases in North America. These differences may be attributed to true differences in genetic susceptibility, or just as likely to differences in urinalysis screening practices, indications for renal biopsy, and possibly other factors. The disease is more common in whites, Asians, and American Indians and less common in blacks both in the United States and in Africa. It is most frequently diagnosed in young adults in the second and third decade of life and there is a male predominance.
Although numerous putative specific environmental or infectious agents have been suggested as the underlying stimulus for IgAN, none has been conclusively confirmed in the majority of patients. Familial clustering and a higher risk in identical twins suggest a role for genetic factors in susceptibility. A variety of genetic polymorphisms associated with susceptibility (and/or progression) in IgAN have been reported with conflicting results. Although IgAN is most commonly a primary (or idiopathic) renal disease, there are well-documented associations with a wide variety of conditions in individual patients (Table 27–1). In many of these conditions, IgA deposition in the glomerulus is not associated with inflammation and a progressive course, and thus, it may be clinically insignificant. This suggests that factors beyond the deposition of IgA or IgA-containing immune complexes must be involved in the pathogenesis of progressive disease in patients with idiopathic IgAN.
Gastrointestinal | Chronic liver disease Celiac disease Inflammatory bowel disease |
Infection | Human immunodeficiency virus Toxoplasmosis Leprosy Yersinia enterocolitica enteritis |
Rheumatic | Ankylosing spondylitis Rheumatoid arthritis Reiter’s syndrome Anterior uveitis |
Pulmonary | Pulmonary hemosiderosis Interstitial pneumonitis |
Malignancy | Lung adenocarcinoma Monoclonal IgA gammopathy Mycosis fungoides |
Dermatologic | Dermatitis herpetiformis Psoriasis |
The precise etiology of IgAN is unknown, but much evidence suggests that a basic abnormality in the IgA molecule itself plays a pivotal role in pathogenesis. Humans produce two isotype subclasses of IgA: IgA1 and IgA2. Plasma cells in the bone marrow, lymph nodes, and spleen mainly produce IgA1 (primarily monomeric), whereas both IgA1 and IgA2 are produced by the plasma cells in the respiratory and gastrointestinal tract (primarily polymeric). Analysis of kidney eluates from patients with IgAN reveals that the glomerular deposits are almost exclusively polymeric IgA1. Furthermore, the hinge region of the IgA1 molecule in patients with IgAN is often abnormal with reduced galactose and/or sialic acid content. The mechanisms responsible for this underglycosylation are unclear, but reduced function of the enzyme responsible for performing this glycosylation may be involved. Nevertheless, the altered glycosylation pattern might favor self-aggregation of IgA1, formation of circulating IgA-containing immune complexes, defective clearance of abnormal IgA1 from the circulation, and increased binding of IgA1 to the extracellular matrix components in the kidney. Once deposited in the kidney, IgA1-containing immune complexes trigger cellular proliferation and enhance the production of proinflammatory cytokines, chemokines, and growth factors. Interleukin (IL)-6 may play a prominent role, but other factors including IL-1, platelet-derived growth factor, tumor necrosis factor, free oxygen radicals, and vascular cell adhesion molecule-1 have also been implicated as modulators of disease activity. Complement activation may be mediated by the alternative or lectin pathways since polymeric IgA1 and aberrantly glycosylated IgA1 are efficient at initiation.
Patients with IgAN present in a variety of ways. Recurrent episodes of painless macroscopic hematuria (brown- or tea-colored urine) are more common in younger patients. These episodes often coincide with, or occur within, 1–2 days of an upper respiratory infection and are referred to as “synpharyngitic.” Macroscopic hematuria may also be temporally related to other acute infections (gastroenteritis or urinary tract infection) and occasionally to strenuous exercise. Dull loin pain may accompany the hematuria, presumably due to renal capsular swelling.
In 30–40% of patients, IgAN is indolent with microscopic hematuria and proteinuria incidentally discovered on urinalysis. This asymptomatic presentation is more common in adults who are diagnosed with the urinary abnormalities at insurance physicals, pregnancy screening, and routine annual checkups. Other less common presentations include the nephrotic syndrome, acute renal failure (which may be the result of crescentic glomerulonephritis, renal tubular obstruction by red blood cells, or acute tubular necrosis), or chronic kidney disease representing long-standing disease that has gone undetected.
There are no specific laboratory tests that distinguish IgAN from other glomerular diseases. The serum creatinine may be normal or elevated at presentation. Hematuria usually dominates the urinalysis. Proteinuria is often present, but the nephrotic syndrome is uncommon (˜10–15% of patients). Urine microscopy typically reveals dysmorphic red blood cells and red blood cell casts indicating bleeding of glomerular origin. Complement levels are normal. Some, but not all IgAN patients have elevated levels of serum IgA (IgA1). Nevertheless, measurement of IgA levels has no diagnostic or prognostic value. Increased IgA levels alone are insufficient to cause disease. This conclusion is supported by observations that other diseases associated with increased serum IgA (ie, HIV, hepatobiliary disease, and IgA myeloma) infrequently result in IgAN.
A kidney biopsy is required for definitive diagnosis of IgAN. Immunofluorescence microscopy demonstrates the pathognomonic finding of dominant or codominant deposits of IgA in a diffuse granular pattern predominantly within the mesangium often with focal paramesangial and subendothelial extension (Figure 27–1). Other immunoglobulins and complement may be codeposited (with less intensity) including IgG, IgM, C3, and λ and κ light chains. Electron microscopy confirms the presence of electron-dense deposits in the mesangium. Light microscopic findings are variable and can reveal mesangial cell proliferation, mesangial expansion, focal or diffuse proliferative glomerulonephritis, crescentic glomerulonephritis, chronic sclerosing glomerulonephritis, and a membranoproliferative glomerulonephritis type I pattern. A subset of nephrotic patients with normal appearing glomeruli by light microscopy may have only prominent visceral epithelial cell foot process effacement on electron microscopy and appear indistinguishable from patients with minimal change disease.
Figure 27–1.
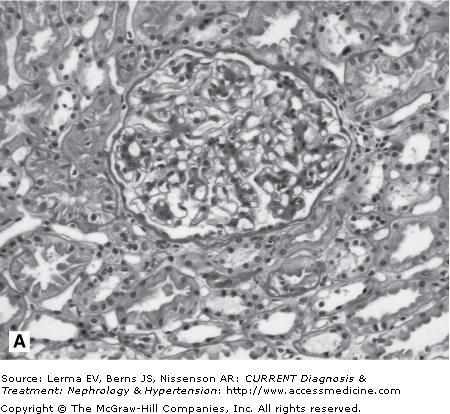
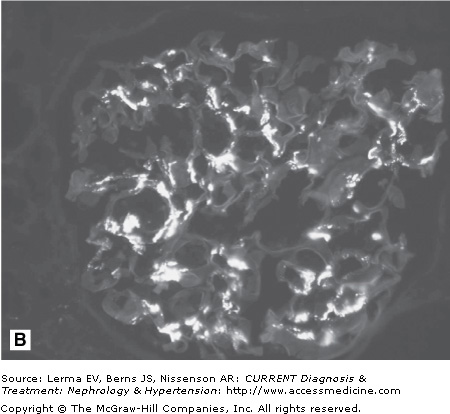
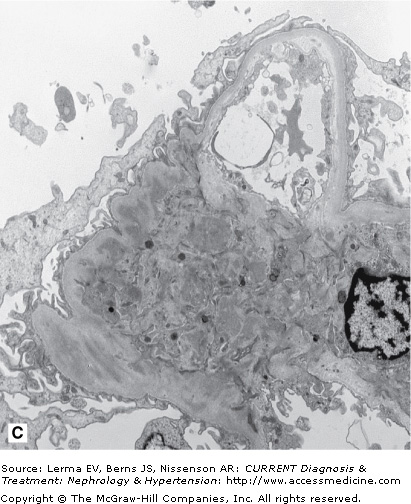
Histology in IgA nephropathy. A: Light micrograph of mesangial glomerulonephritis typical of immunoglobulin A nephropathy. There are segmental areas of increased mesangial matrix and cellularity. B: Direct immunofluorescence microscopy demonstrating large, globular mesangial IgA deposits. C: Electron micrograph demonstrating electron-dense deposits in the mesangium.









