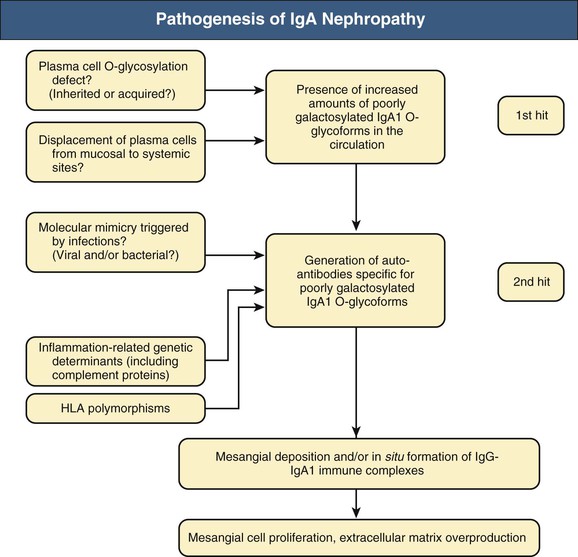
John Feehally, Jürgen Floege IgA nephropathy (IgAN) is a mesangial proliferative glomerulonephritis characterized by diffuse mesangial deposition of IgA. Also called Berger disease, IgAN was first recognized in 1968 by Jean Berger when immunofluorescence techniques were introduced for the study of renal biopsy specimens. IgAN is unique among glomerular diseases in being defined by the presence of an immune reactant rather than by any other morphologic feature on renal biopsy, and the light microscopy changes are variable. IgAN is the most prevalent pattern of glomerular disease seen in most Western and Asian countries where renal biopsy is widely practiced. The term benign recurrent hematuria was previously used for IgAN, but it is now known that IgAN is an important cause of end-stage renal disease (ESRD). It is likely that IgAN is not a single entity but rather a common response to various injurious mechanisms. Henoch-Schönlein purpura (HSP) is a small-vessel vasculitis affecting the skin, joints, gut, and kidneys that predominantly affects children. It is defined by tissue deposition of IgA. HSP was described separately by Schönlein in 1837 and Henoch in 1874. Typically, there is clinical involvement in the skin, gut, and kidneys. The nephritis associated with HSP is also characterized by mesangial IgA deposition; indeed, the renal histologic features of Henoch-Schönlein (HS) nephritis are indistinguishable from those of IgAN. HS nephritis is differentiated from IgAN by the extrarenal manifestations. Although infective episodes precede HSP in up to 50% of cases and usually precipitate macrohematuria episodes in young IgAN patients, no evidence indicates a role for any specific antigen. The clinical association of visible hematuria with upper respiratory tract infection in IgAN indicates that the mucosa may be a site of entry for foreign antigens. An infectious source has long been suspected, and there have been occasional reports of IgAN in association with microbial infection, both bacterial (e.g., Campylobacter, Yersinia, Mycoplasma, Haemophilus) and viral (e.g., cytomegalovirus, adenovirus, coxsackievirus, Epstein-Barr virus). A severe form of IgAN, which may be crescentic, has been reported in association with severe staphylococcal infection. No organism, however, has been consistently implicated by the finding of microbial antigen in glomerular deposits in typical cases of IgAN. In addition there is evidence for a mucosal hyperresponsiveness to a variety of food antigens in IgAN patients.1 The mesangial IgA may thus represent a common immune response to a variety of foreign antigens. Another common feature of this immune response is that the deposited IgA is undergalactosylated, and that the occurrence of this type of IgA in the circulation may elicit an autoimmune IgA- and IgG-response (see later discussion). The regular recurrence of IgAN and HS nephritis after renal transplantation strongly implies an abnormality in the host IgA immune system (see Chapter 108). IgA is the most abundant immunoglobulin in the body and is chiefly concerned with mucosal defense. It has two subclasses, IgA1 and IgA2. Mucosal antigen challenge provokes polymeric IgA (pIgA) production by plasma cells of the mucosa-associated lymphoid tissue; the pIgA is then transported across epithelium into mucosal fluids, where it is released after coupling to secretory component as secretory IgA (sIgA). The function of circulating IgA is less clear; it is bone marrow derived and mostly monomeric IgA1 (mIgA1). Circulating IgA1 is cleared by the liver through hepatocyte asialoglycoprotein receptors and Kupffer cell Fcα receptors. The mesangial IgA in IgAN is predominantly pIgA1. The clinical association with mucosal infection or superantigens from Staphylococcus aureus originally suggested that the mesangial pIgA1 comes from the mucosal immune system. In IgAN, however, pIgA1 production is downregulated in the mucosa and upregulated in the bone marrow. Moreover, the pIgA response to systemic immunization with common antigens is increased, whereas the response to mucosal immunization is impaired. Impaired mucosal IgA responses allowing enhanced antigen challenge to the marrow could be the primary abnormality in IgAN, although this remains unproven. Similarly attractive, yet still unproven, is the hypothesis that some mucosal IgA-producing plasma cells translocate to the bone marrow in IgAN; this could also explain the particular glycosylation of serum IgA in IgAN. Tonsillar pIgA1 production is also increased, although IgAN can occur after tonsillectomy, and the tonsil is a very minor source of IgA production compared with the mucosa or marrow. There are reports of sIgA in the mesangium, but this finding is not easily explained by current pathogenic concepts of IgAN. Hepatic clearance of IgA is reduced, possibly because of the altered molecular characteristics of IgA in IgAN (see later discussion). Serum IgA levels are increased in one third of patients with IgAN and HSP. There are elevations in both mIgA and pIgA. However, high serum IgA itself is not sufficient to cause IgAN. High circulating levels of monoclonal IgA in myeloma or polyclonal IgA, in acquired immunodeficiency syndrome (AIDS), only infrequently provoke mesangial IgA deposition. Circulating macromolecular IgA is characteristic of IgAN. It is often described as IgA immune complexes, although the antigen is only rarely identified. There are circulating IgA rheumatoid factors (IgA against constant domain of IgG) in 30% of patients with IgAN and 55% of those with HSP. Studies in vitro indicate that IgA production by mononuclear cells is exaggerated in IgAN, and that these cells show abnormal patterns of cytokine production. However, the direct relevance of these observations to events in vivo is uncertain. IgA1 carries distinctive O-linked sugars at its hinge region; IgA2 has no hinge and carries no such sugars. There is good evidence that circulating IgA1 in IgAN and HS nephritis has abnormal O-linked hinge-region sugars with reduced galactosylation because of altered IgA production in lymphocytes of patients with IgAN.1 Some data indicate defective function of the relevant glycosyltransferases that O-glycosylate IgA1, possibly with a genetic basis. Other findings suggest that the primary abnormality may be that IgA of mucosal type, which has glycosylation patterns different from serum IgA1, reaches the circulation, for example, by translocation of mucosal lymphocytes to the bone marrow. The latter is consistent with experiments, where immortalized lymphocytes from IgAN patients continue to produce dimeric and polymeric IgA with altered galactosylation in vitro.2 Mesangial IgA1 in IgAN has the same abnormalities of O-glycosylation.3,4 The altered glycosylation may lead to IgG anti-IgA autoantibodies and promote mesangial IgA1 deposition by predisposing to the formation of circulating IgA1 immune complexes or by directly modifying IgA1 interactions with matrix proteins and mesangial cell or monocyte Fc receptors.5 It may also impair IgA1 clearance by inhibiting IgA1 interactions with hepatic and circulating myeloid cell IgA receptors. Figure 23-1 summarizes some key elements involved in the pathogenesis of IgAN. Polymeric IgA deposition in the mesangium is typically followed by mesangial proliferative glomerulonephritis (GN). In animal models, co-deposition of IgG and complement is necessary for inflammation, but this is not mandatory in human disease. Complement deposits are usually C3 and properdin without C1q and C4. There may be complement activation via the mannose-binding lectin pathway. The extent to which IgA engages inflammatory cells in the circulation and especially in the kidney will determine the intensity of inflammation. Fc receptors for IgA (Fcα receptors) on myeloid and mesangial cells may play a key role.6 The mechanisms of mesangial proliferative GN have been studied in detail in animal models, particularly anti–Thy 1 nephritis in the rat. These studies have shown the key role of cytokines and growth factors in mesangial cell proliferation, particularly the B and D isoforms of platelet-derived growth factor7 (PDGF) and in the subsequent matrix production and sclerosis, particularly transforming growth factor-β (TGF-β). Studies of renal biopsy specimens in human IgAN also support a role for PDGF and TGF-β. These mechanisms are not unique to IgAN but are likely involved in all forms of mesangial proliferative GN, including those without IgA deposition. Animal IgA does not have the same characteristics as human IgA1, and some animals also have IgA clearance mechanisms distinct from those in humans. Therefore, animal models, even if they provoke mesangial IgA deposits, are not particularly informative about the mechanisms that underlie human mesangial pIgA1 deposition, although they have provided many insights into events after IgA deposits have developed. There is no animal model in existence for HSP. Despite some differences in age at onset and natural history of IgAN and HS nephritis,8 there is much evidence to support a close pathogenetic link between the two conditions, and the outcome of the two diseases may be similar.9 Monozygotic twins who developed IgAN and HSP, respectively, at the same time have been described. The evolution of IgAN into HSP in the same patient is described in both adults and children, and HSP patients with ESRD receiving a renal transplant may experience recurrent disease in the form of IgAN. Many of the abnormalities of IgA production and handling reported in IgAN are also detected in HSP. IgA nephropathy is the most prevalent pattern of glomerular disease in most countries where renal biopsy is widely used as an investigative tool. Its estimated frequency is at least 2.5 cases per year per 100,000 adults.10 However, the striking geographic variation has been associated with the presence of particular gene alleles that protect from IgAN11 (Fig. 23-2). This racial predisposition is maintained in other locations; in the United States, for example, IgAN is less common in Blacks than in Caucasians of European origin. Perceived prevalence of IgAN may also be influenced by attitudes to the investigation of microhematuria. A country with an active program of routine urine testing will inevitably identify more individuals with urine abnormalities, but IgAN will be identified only if renal biopsy is performed. Even then, the prevalence of IgAN will be underestimated; a study of kidney donors suggests that the prevalence of IgAN with mesangial proliferative changes and glomerular C3 deposits in the general population in Japan may be 1.6%.12 This suggests that the vast majority of IgAN patients never come to medical attention and spontaneously remit. In children, HSP is usually diagnosed on clinical grounds without biopsy confirmation of tissue IgA deposition. Transient urine abnormalities are common in the acute phase. However, only those with persistent urine abnormalities or with more overt renal disease will come to renal biopsy. Therefore, the incidence of HS nephritis is almost certainly underestimated, with many unidentified mild and transient cases. There is no information on geographic variations in HSP. Urine abnormalities increase in frequency among the relatives of IgAN patients, although only in a few pedigrees is IgAN found in multiple generations. One large pedigree has been described in Kentucky, and other large families have been found in Italy and Canada. However, more than 90% of all cases of IgAN appear to be sporadic. Large worldwide genome-wide association studies have identified genetic modulators that seem to affect the prevalence of sporadic IgAN and modulate its course.11–13 Variations in major histocompatibility gene loci (HLA-DR, -DQ, -DP and HLA-B) have consistently been identified. Other gene loci are less consistent and include inflammatory mediators (tumor necrosis factor and α-defensin) and gene loci affecting complement factor H.14 The wide range of clinical presentations of IgAN varies in frequency with age (Fig. 23-3). No clinical pattern is pathognomonic of IgAN. In populations of Caucasian descent, IgAN is more common in males than females by a ratio of 3 : 1, whereas the ratio approaches 1 : 1 in most Asian populations. In 40% to 50% of patients with IgAN, the clinical presentation is episodic macroscopic hematuria, most frequently in the second decade of life. The urine is usually brown rather than red, and clots are unusual. There may be loin pain caused by renal capsular swelling. Hematuria usually follows intercurrent mucosal infection, typically in the upper respiratory tract (the term synpharyngitic hematuria has been used) or occasionally in the gastrointestinal tract. Hematuria is usually visible within 24 hours of the onset of the symptoms of infection, differentiating it from the 2- to 3-week delay between infection and subsequent hematuria in postinfectious (e.g., poststreptococcal) GN. The macroscopic hematuria resolves spontaneously over a few days. Microscopic hematuria persists between attacks. Most patients have only a few episodes of frank hematuria, which become less frequent and resolve over a few years or sooner. Such episodes may be associated with acute kidney injury (AKI). Asymptomatic urine testing identifies 30% to 40% of patients with IgAN in most reported series. Microhematuria with or without proteinuria (usually <2 g/24 h) is noted. The number of patients identified in this way will depend on local attitudes to urine screening as well as on the use of renal biopsy in patients with isolated microscopic hematuria. Most patients with IgAN are asymptomatic and would be identified only when the urine is tested. It is rare for proteinuria to occur without microscopic hematuria. Whereas nephrotic-range proteinuria may occur, in particular in the presence of uncontrolled hypertension, full-blown nephrotic syndrome is uncommon, occurring in only 5% of all patients with IgAN. Nephrotic syndrome may occur early in the course of the disease, with minimal glomerular change or with active mesangial proliferative GN. Alternatively, it may occur as a late manifestation of advanced chronic glomerular scarring. Although uncommon in IgAN (<5% of all cases), one study reports that AKI may be the presentation in up to 27% of patients older than 65 years.15 AKI develops by three distinct mechanisms. There may be acute severe immune and inflammatory injury with necrotizing GN and crescent formation (crescentic IgAN); this may be the first presentation of IgAN, or it may be superimposed on established, less aggressive disease. Rapid deterioration in IgAN in pregnancy may be caused by crescentic transformation. Alternatively, AKI can occur with mild glomerular injury when heavy glomerular hematuria leads to tubule occlusion by red blood cells (RBCs). Third, especially in elderly patients, chronic IgAN will predispose to AKI from a variety of incidental renal insults (see Chapter 69). Some patients already have renal impairment and hypertension when they are first diagnosed with IgAN. These patients tend to be older, and they probably have longstanding disease that previously remained undiagnosed because the patient neither had frank hematuria nor underwent routine urinalysis. Hypertension is common, as in other chronic glomerular disease; accelerated hypertension occurs in 5% of patients. Mesangial IgA deposition is a frequent finding in autopsy studies in chronic liver disease. Although particularly associated with alcoholic cirrhosis, IgA deposition can occur in other chronic liver disease, including that caused by hepatitis B and schistosomiasis. It is thought to result from impaired clearance of IgA by the Kupffer cells, which express Fcα receptors, and hepatocytes, which express the asialoglycoprotein receptor. Clinical evidence of renal disease is more common than previously appreciated, and patients occasionally develop ESRD. A number of case reports have associated IgAN with human immunodeficiency virus and AIDS. It is not clear whether the polyclonal increase in serum IgA, which is a feature of AIDS, is the predisposing factor. There are case reports associating IgAN with many other conditions, particularly with a number of immune and inflammatory diseases (Table 23-1). Their relationship to abnormalities of the IgA immune system is not always clear, and some may represent the coincidental development of unrelated but relatively common conditions. Table 23-1 Diseases reported in association with IgA nephropathy: common, reported, and rare. Rare associations have been made in one or two reported cases only. In a disease as common as IgA nephropathy, it is therefore uncertain whether these are truly related. HIV, Human immunodeficiency virus. * Behçet syndrome: systemic vasculitis typified by orogenital ulceration and chronic uveitis. † Takayasu arteritis: systemic vasculitis involving the aorta and its major branches, most often found in young women. ‡ Wiskott-Aldrich syndrome: X-linked disorder in which increased serum IgA is associated with the triad of recurrent pyogenic infection, eczema, and thrombocytopenia. Although most prevalent in the first decade of life, HSP may occur at any age. A palpable purpuric rash, which may be recurrent, occurs on extensor surfaces (Fig. 23-4). There may be polyarthralgia (usually without joint swelling) and abdominal pain caused by gut vasculitis. This may be severe, with bloody diarrhea if intussusception develops. In practice, the diagnosis is made by clinical criteria in the great majority of children, in whom HSP is a self-limiting illness. In adults, clinical features include purpura, arthritis, and gastrointestinal symptoms in 95%, 60%, and 50% of patients, respectively.16
IgA Nephropathy and Henoch-Schönlein Nephritis
Definitions
Etiology and Pathogenesis
IgA Immune System
IgA Glycosylation
Glomerular Injury After IgA Deposition
Animal Models of IgA Nephropathy
Relationship Between IgA Nephropathy and Henoch-Schönlein Purpura
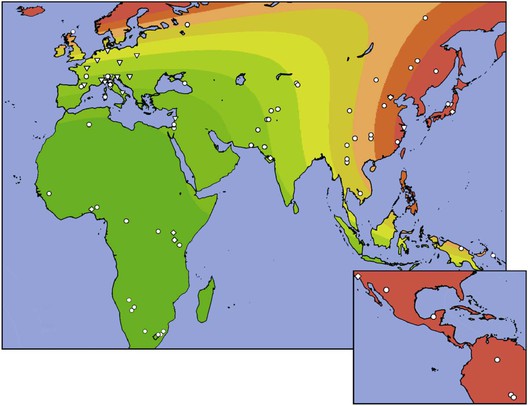
Epidemiology
Genetic Basis of IgA Nephropathy
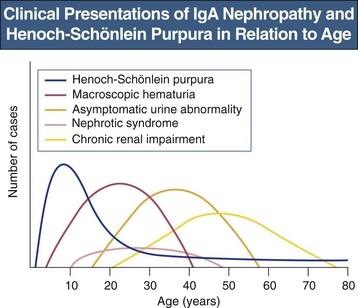
Clinical Manifestations
IgA Nephropathy
Macroscopic Hematuria
Asymptomatic Hematuria and Proteinuria
Proteinuria and Nephrotic Syndrome
Acute Kidney Injury
Chronic Kidney Disease
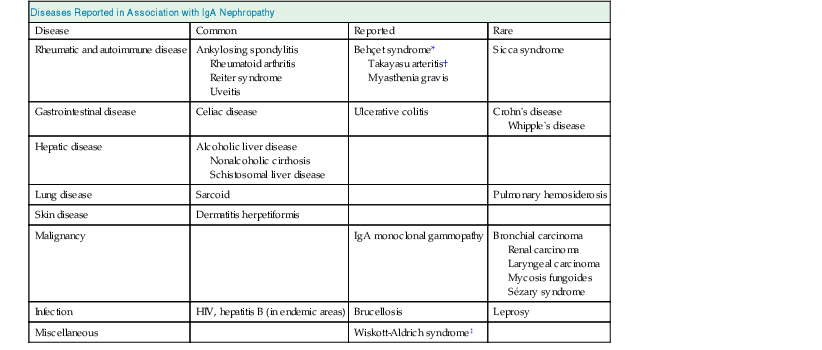
Clinical Associations with IgA Nephropathy
Diseases Reported in Association with IgA Nephropathy
Disease
Common
Reported
Rare
Rheumatic and autoimmune disease
Ankylosing spondylitis
Rheumatoid arthritis
Reiter syndrome
Uveitis
Behçet syndrome*
Takayasu arteritis†
Myasthenia gravis
Sicca syndrome
Gastrointestinal disease
Celiac disease
Ulcerative colitis
Crohn’s disease
Whipple’s disease
Hepatic disease
Alcoholic liver disease
Nonalcoholic cirrhosis
Schistosomal liver disease
Lung disease
Sarcoid
Pulmonary hemosiderosis
Skin disease
Dermatitis herpetiformis
Malignancy
IgA monoclonal gammopathy
Bronchial carcinoma
Renal carcinoma
Laryngeal carcinoma
Mycosis fungoides
Sézary syndrome
Infection
HIV, hepatitis B (in endemic areas)
Brucellosis
Leprosy
Miscellaneous
Wiskott-Aldrich syndrome‡
Henoch-Schönlein Purpura
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


IgA Nephropathy and Henoch-Schönlein Nephritis
Chapter 23