Hypertension in Chronic Kidney Disease
Peter N. Van Buren
Robert D. Toto
INTRODUCTION
The prevalence of hypertension in patients with chronic kidney disease (CKD) exceeds that of the general population. Although hypertension is believed to be the etiology of kidney disease in many of these patients, hypertension is often the consequence of kidney disease stemming from any etiology. The pathophysiology of hypertension in CKD is related to multiple factors, including expanded extracellular volume from sodium retention, activation of the sympathetic nervous system (SNS) and the renin-angiotensin-aldosterone system (RAAS), and imbalances in vasoconstrictor and vasodilator substances that regulate peripheral vascular resistance. Untreated or poorly controlled hypertension in CKD patients is associated with adverse outcomes including deterioration of renal function, development of left ventricular hypertrophy (LVH), and increased mortality. Recent clinical trials have investigated the role of various antihypertensive treatments and blood pressure targets in preventing these outcomes. The purpose of this chapter is to review the epidemiology, pathophysiology, and management of hypertension in patients with CKD. Because diabetic nephropathy is the leading cause of end-stage renal disease (ESRD) in the United States, the unique aspects of its pathophysiology and the treatment of hypertension in this setting warrant a separate discussion. Although mentioned briefly herein, the reader is referred to a recent extensive review of this topic.1
EPIDEMIOLOGY
Chronic Kidney Disease
Hypertension is a prevalent comorbidity associated with CKD. The prevalence of hypertension in the overall U.S. population, according to the National Health and Nutrition Examination Survey (NHANES) data from 2007 to 2008, is 29%.2 Data published by the United States Renal Data System (USRDS) according to NHANES data collected from 1999 to 2006 indicate that the prevalence of hypertension in the non-CKD population is 23.3%.3 In contrast, the prevalence of hypertension in the CKD population is much higher. The prevalence of hypertension increases with each stage of CKD and is estimated to be 35.8%, 48.1%, 59.9%, and 84.1% in patients with stage I, II, III, and IV/V CKD, respectively. Although the awareness of hypertension among patients with stage III and IV CKD was similar to that in the non-CKD population, the failure to control hypertension (defined as blood pressure [BP] 130/80 mm Hg for those with CKD and 140/90 mm Hg for those without CKD) is higher among the CKD patients.
The Chronic Renal Insufficiency Cohort (CRIC) is a National Institutes of Health (NIH) sponsored prospective observational study among 3,612 patients with an estimated glomerular filtration rate (GFR) of 20 to 70 mL per minute designed to better understand factors responsible for CKD progression and cardiovascular disease.4 Although the sample population for CRIC is smaller than NHANES, the detailed ascertainment of individual demographics, comorbidities, medication use, and laboratory analysis provides useful information for understanding the relationship between CKD and hypertension control in a population that is established in the health care system. In this population, 85.7% of patients are hypertensive based on the definition of a BP > 140/90 mm Hg or with the use of an antihypertensive medication. The percentage of patients from CRIC that are aware of their diagnosis of hypertension (98.9%) and who are treated for hypertension (98.3%) is higher than in the NHANES data. However, the control of hypertension in CRIC patients is still suboptimal, with 67.1% having BP > 130/80 mm Hg and 46.7% having BP > 140/90 mm Hg. The CRIC data indicate that older age, African American race, and a greater amount of proteinuria are risk factors for a failure to control BP to either 140/90 or 130/80 mm Hg.5 Overall, the epidemiologic evidence from NHANES and CRIC identifies that hypertension is a significant burden for patients with CKD and the health care providers responsible for managing these patients.
The long-term consequences of uncontrolled hypertension highlight the significance of this disease in CKD patients. Studies have reported that elevated systolic BP increases the incidence of CKD,6,7 the progression of CKD,8 and the incidence of ESRD.9 Furthermore, hypertension is reported to be the second leading cause of ESRD in the
United States based on USRDS data.10 However, it has been difficult to establish the independent effect of BP in CKD from effects related to the degree of baseline proteinuria.11
United States based on USRDS data.10 However, it has been difficult to establish the independent effect of BP in CKD from effects related to the degree of baseline proteinuria.11
Elevated BP is also a risk factor for cardiovascular events, including stroke and myocardial infarction among CKD patients; however, the exact relationship between BP and outcomes is not consistent among studies. A J-shaped relationship between cardiovascular morbidity and BP has been shown,12 suggesting that the highest risk for an outcome occurs at the highest and lowest BP, whereas the lowest risk for an outcome occurs at an intermediate BP. In one longitudinal study of patients with stage III and IV CKD, systolic BP >130 mm Hg was a predictor for an incident stroke; however, those with systolic BP < 120 mm Hg had a greater risk than those with a systolic BP between 120 and 129 mm Hg.12 In contrast, a post hoc analysis of the Perindopril Protection Against Recurrent Stroke Study (PROGRESS), a prospective randomized placebo controlled trial of the effects of perindopril on stroke among patients with a prior history of cerebrovascular disease, CKD patients had a reduced risk for a recurrent stroke across all strata of systolic BP. Moreover, there was no increase in the risk for recurrent stroke in those who achieved a systolic BP <120 mm Hg compared to higher achieved BP levels.13
When considering mortality as an outcome, some observational data confirm the association with low BP and events.14,15 One study showed an increased mortality risk in subjects with baseline systolic BP in the lowest quartile (<133 mm Hg) compared to the other quartiles, and another study showed the highest mortality risk with a systolic BP < 110 mm Hg and >180 mm Hg in a cohort of CKD patients inclusive of both diabetic and nondiabetic CKD. The effect from the latter study was strongest in older patients with advanced CKD and without proteinuria, thus limiting the generalizability of this finding.
In summary, observational studies demonstrate an increased risk for cardiovascular morbidity and mortality at BP levels considered hypertensive for the general population. However, it is unclear if an aggressive reduction of BP translates into decreased cardiovascular morbidity and mortality in the CKD population. The evidence from randomized clinical trials on specific BP targets is discussed in the treatment section of the chapter.
Ambulatory Blood Pressure Measurements and Chronic Kidney Disease
Although CKD patients frequently have BP measured in an office setting, it is important to recognize some of the limitations that can arise in this context. Of utmost importance to the topic of hypertension is the relationship between home and clinic BP measurements. Home and ambulatory measurements better predict the presence of end organ damage such as proteinuria compared to clinic measurements.16 Ambulatory BP measurements also predict which CKD patients with an elevated clinic BP have a greater risk for progression to ESRD or reaching the composite outcome of ESRD or death.17 Thus, a comprehensive ascertainment of BP burden requires the consideration of more than measurements obtained in the office.
Hemodialysis Patients
ESRD patients on hemodialysis (HD) have an annual mortality rate close to 20%, with cardiovascular disease and infections accounting for the highest percentage of deaths.18 Although the prevalence of hypertension in the HD population is near 90%,19 a target BP to improve outcomes has yet to be identified. Early epidemiologic studies showed that for BP measurements obtained in the HD unit, low systolic BP and systolic BP in excess of 200 mm Hg were associated with the highest mortality, particularly in older patients and diabetics.20,21 However, it has also been shown that uncontrolled hypertension with systolic BP in excess of 140 mm Hg results in the increased incidence of LVH, de novo ischemic heart disease, and de novo cardiac failure.22 It must be considered that low systolic BP can be a manifestation of decreased cardiac output, resulting from the structural and functional consequences of long-standing uncontrolled hypertension, which would explain its association with increased mortality.
Similar to pre-ESRD CKD patients, the timing and location of BP measurements are also important considerations in HD patients. Home and ambulatory BP measurements during the interdialytic time period, in comparison to individual HD-unit measurements, are better predictors of mortality.23 The significance of BP changes during HD treatments has also been recently investigated. Intradialytic hypertension, increases in BP from pre- to post-HD, has been associated with increased short-term (6 month) morbidity and mortality in prevalent HD patients and decreased 2-year survival in incident HD patients.24,25 There is evidence that mechanisms responsible for the phenomenon include extracellular volume overload26 or increased vasoconstriction related to intradialytic endothelin-1 surges as a manifestation of endothelial cell dysfunction.27,28,29 Patients with intradialytic hypertension have also been shown to have increased ambulatory blood pressure and greater impairment in underlying endothelial cell function during the interdialytic time period.30,31 Additional mechanisms that have been proposed, but that have yet to be confirmed, include increased activity of the RAAS and SNS, changes in electrolytes during HD, and removal of antihypertensive medications during the course of HD.32
PATHOPHYSIOLOGY
The etiology of hypertension in CKD is multifactorial and related to both increases in cardiac output and increased peripheral vascular resistance (Table 40.1). Positive sodium balance can affect either component (Fig. 40.1), and achieving sodium balance remains a primary target of managing hypertension in patients with CKD. Multiple other mechanisms, including activation of the SNS and RAAS, endothelial cell dysfunction related to imbalances in vasodilator and vasoconstrictor substances, and increased oxidative stress, can modify the effects of each other and result in hypertension, particularly when these systems are disrupted in the context of CKD. A comprehensive summary of these processes is outlined in Figure 40.2.
TABLE 40.1 Etiology of Hypertension in Chronic Kidney Disease | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
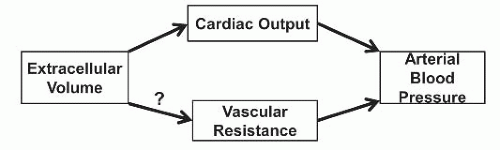 FIGURE 40.1 Blood pressure is directly related to cardiac output and vascular resistance. Extracellular volume increases cardiac output and, potentially, vascular resistance in patients with chronic kidney disease and end-stage renal disease. The achievement of euvolemia and a reduction in vascular resistance remain the primary target of blood pressure reduction in these patient populations. |
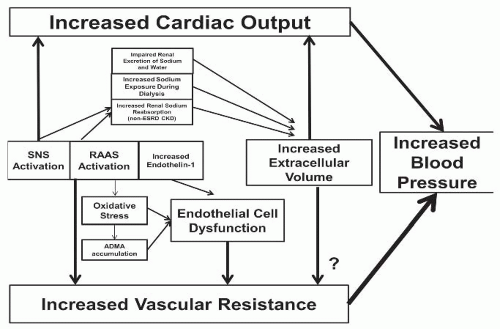 FIGURE 40.2 Both increased cardiac output and increased vascular resistance contribute to increased blood pressure in patients with chronic kidney disease (CKD) and end-stage renal disease (ESRD). Increased cardiac output results primarily from increased extracellular volume. Activation of the sympathetic nervous system (SNS) and the renin-angiotensin-aldosterone system (RAAS) contributes to renal sodium reabsorption in non-ESRD CKD patients. In both CKD and ESRD patients, the decreased renal excretion of sodium and water from the decreased glomerular filtration rate also contributes to increased extracellular volume. Finally, in ESRD patients on hemodialysis, the transfer of sodium from the dialysate to the plasma can promote thirst and interdialytic weight gain. Vascular resistance can be increased by the activation of the SNS and the RAAS, as well as by enhanced vasoconstriction caused by endothelial cell dysfunction. Increased RAAS activity increases angiotensin (Ang) II, which binds to receptors on vascular smooth muscle cells and causes vasoconstriction. Ang II also activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and increases oxidative stress, which is believed to be responsible for the increase in the inhibitor of endothelial nitric oxide synthase, asymmetric dimethylarginine. Endothelial cell dysfunction refers to an imbalance in mediators released from the endothelial cells of which one of the results is increased vasoconstriction. An increase in asymmetric dimethylarginine (ADMA) interferes with the production of the vasodilator nitric oxide (NO). Reactive oxidative species also combine with NO to form peroxynitrite and to prevent NO that has already been produced from binding to its receptor. In the context of impaired production and function of NO, there is less opposition to the vasoconstrictive effects of endothelin (ET)-1. Extracellular volume overload has varying effects on vascular resistance, but there is evidence that there is a delayed increase in vascular resistance that follows volume-induced increases in blood pressure and acts to sustain hypertension. |
Basic Concepts
BP in humans is determined both by the cardiac output and by peripheral vascular resistance. Cardiac output is dependent on the intravascular component of the extracellular space as well as the heart rate; and peripheral resistance is dependent on functions of the vascular endothelial and smooth muscle cells in response to the actions of various vasoactive mediators (both vasoconstrictors and vasodilators). Although alteration of either cardiac output or vascular resistance would initially be expected to affect BP in a concordant direction, a healthy kidney can adapt to short-term changes in BP to restore normotension. An increase in renal sodium excretion is the expected response to increased BP in healthy individuals. This pressure natriuresis allows for the restoration of extracellular volume and BP following an increase in cardiac output. However, in CKD, this homeostatic mechanism is impaired, and the kidney fails to sufficiently excrete sodium loads. In CKD and ESRD, it is hypothesized that an increase in cardiac output initiates the increase in BP, but ultimately, an increase in vascular resistance sustains the BP elevation.33,34 Accordingly, addressing both volume status and the degree of peripheral vasoconstriction are necessary to control BP in these patient populations.
Sodium Balance and Volume Overload
Sodium balance is an important aspect in BP control in CKD and ESRD. Because of the kidney’s ability to excrete sodium, healthy individuals can tolerate large amounts of sodium intake without significant increases in BP.35 However, in the presence of kidney disease, BP is highly dependent on extracellular volume. Rats that have undergone 70% renal ablation develop severe hypertension on a high sodium diet, but hypertension is completely prevented in these animals while on a low sodium diet.36 A lower hematocrit found in the animals on a high sodium diet suggested that extracellular volume overload was present and likely the mechanism responsible for the increased BP. Another experiment with a renal ablation model demonstrated that increased cardiac output is responsible for the initial increase in BP during acute salt loading, but increased peripheral resistance (which occurs after BP has already become increased) is responsible for the maintenance of elevated BP even after cardiac output has returned to normal.37
Chronic Kidney Disease Patients Not on Hemodialysis
Because CKD patients are limited in the amount of renal sodium excretion, BP and sodium balance are interrelated. Among CKD patients, those with more severe renal impairment experience greater increases in BP in response to a sodium load. Although the pressure natriuresis curve serves to maintain sodium balance in healthy individuals, CKD patients require much higher BP increases than healthy individuals to augment their renal sodium excretion. Similarly, the degree of blood volume expansion required to induce a natriuresis is higher in patients with severe renal impairment compared to those with moderate renal impairment. Overall, these findings show that the effects on BP related to sodium intake (“salt sensitivity”) are augmented with the progression of renal disease.38 During the early stages of CKD, despite suboptimal renal sodium excretion, some patients remain normotensive despite an increase in cardiac output because of a reduction in peripheral resistance.33 Although BP is sustained in the normotensive range, these patients, who are already limited in their sodium excretion because of reductions in GFR, fail to induce the necessary pressure natriuresis to re-establish sodium balance. Consequently, further increases in sodium loading result in increased cardiac output, peripheral resistance, and BP.
When subjects with CKD increase dietary sodium ingestion from 20 to 120 mEq per day, they experience an increase in BP that is not seen even in healthy subjects who increase their dietary sodium ingestion to levels as high as 1200 mEq per day.39 Although both groups have a similar suppression of RAAS at moderate amounts of sodium ingestion, plasma renin activity (PRA) and angiotensin (Ang) II decreased drastically in healthy subjects during periods of high sodium ingestion (1200 mEq per day).39 Although vascular resistance could not be measured directly in this study, increased vasoconstriction in the CKD subjects appeared to be the likely mechanism responsible for the increased BP and the reduced distribution of fluid into the interstitial space. Even under clinical conditions of pharmacologic RAAS blockade, salt balance has an important role in BP. In nondiabetic proteinuric subjects with creatinine clearance ranging from 33 to 110 mL per minute ingesting a low sodium diet, a reduction in BP and proteinuria during the chronic administration of fixed doses of angiotensin converting enzyme (ACE) inhibitors is reversed during periods of increased dietary sodium ingestion.40 As further evidence of the importance of sodium intake in relation to BP and proteinuria in CKD patients, the coadministration of hydrochlorothiazide during a high salt intake period reduces both BP and proteinuria back to values seen during the low sodium diet.
Chronic Kidney Disease On Hemodialysis
In ESRD patients with little or no renal sodium excretion, sodium removal through ultrafiltration during HD is the primary means to maintain extracellular volume status because a pressure natriuresis is not possible. Consequently, compared to healthy controls, HD patients have a significantly higher cardiac output.41 However, the presence of hypertension among HD patients is also highly dependent on elevations in vascular resistance.41 Extracellular volume potentially impacts both of these parameters to influence BP. When subjected to sodium loading, HD patients typically respond with an increase in both BP and cardiac output. The pattern of vascular resistance following sodium loading is more variable, but increases typically do not occur until after the elevated cardiac output has already led to an
increase in BP.34,42 In between HD treatments, during the interdialytic period, HD patients gradually gain weight with their routine dietary intake of sodium and water. The BP patterns during a typical interdialytic period show rhythmic oscillations superimposed on a general linear increase in BP over time.43 This pattern is modified by interdialytic weight gain such that greater weight gain is associated with an increase in the slope of BP rise.44 Subsequently, greater increases in interdialytic weight gain have been associated with increased pre-HD systolic BP at the next HD treatment.45 However, that increased interdialytic weight gain is also associated with a greater reduction in BP during the course of that treatment (likely as a response to the ultrafiltration required to remove the interdialytic fluid gain).45 Such evidence supports the hypothesis that extracellular volume (through either increases in cardiac output or vascular resistance) is primarily responsible for hypertension in this population. Consequently, one HD center has described a 98% success rate in withdrawing antihypertensive medications while using the ultrafiltration that can be achieved during a cumulative weekly dialysis time of 24 hours.46 Thus, recognition and successful attainment of a patient’s dry weight can facilitate the initial BP management in most, but not all, HD patients.
increase in BP.34,42 In between HD treatments, during the interdialytic period, HD patients gradually gain weight with their routine dietary intake of sodium and water. The BP patterns during a typical interdialytic period show rhythmic oscillations superimposed on a general linear increase in BP over time.43 This pattern is modified by interdialytic weight gain such that greater weight gain is associated with an increase in the slope of BP rise.44 Subsequently, greater increases in interdialytic weight gain have been associated with increased pre-HD systolic BP at the next HD treatment.45 However, that increased interdialytic weight gain is also associated with a greater reduction in BP during the course of that treatment (likely as a response to the ultrafiltration required to remove the interdialytic fluid gain).45 Such evidence supports the hypothesis that extracellular volume (through either increases in cardiac output or vascular resistance) is primarily responsible for hypertension in this population. Consequently, one HD center has described a 98% success rate in withdrawing antihypertensive medications while using the ultrafiltration that can be achieved during a cumulative weekly dialysis time of 24 hours.46 Thus, recognition and successful attainment of a patient’s dry weight can facilitate the initial BP management in most, but not all, HD patients.
Therefore, HD treatment with adequate ultrafiltration should normalize BP in many HD patients. However, the basis for this practice assumes that interdialytic sodium and fluid intake is not excessive. An insufficient time on HD needed to completely restore normal extracellular fluid volume and achieve dry weight without inducing symptomatic hypotension limits this practice. A cross-sectional study found that major differences between ultrafiltration-sensitive and ultrafiltration-resistant HD patients were the pre-and post-HD atrial natriuretic peptide (ANP) levels between groups.47 The ultrafiltration-sensitive group had reductions in ANP during HD (as well as lower pre-HD ANP), whereas the higher pre-HD ANP levels in the ultrafiltration-resistant group persisted despite similar amounts of ultrafiltration, suggesting that this group had remaining extracellular volume contributing to the elevated BP. It should be recognized, however, that extracellular volume overload may not always manifest itself overtly. In some cases, patients may have additional extracellular volume despite appearing euvolemic on clinical exam. The Dry Weight Reduction in Hypertensive Hemodialysis Patients (DRIP) study was a randomized clinical trial in which hypertensive HD subjects were randomized to either continue their current HD and ultrafiltration prescription or have their dry weight challenged during each HD treatment over several weeks by 0.1 kg per 10 kg dry weight until symptoms developed. The subjects randomized to additional ultrafiltration demonstrated significant decreases in ambulatory systolic BP after 4 weeks and 8 weeks.48
The dialysate used during HD is another important factor that may contribute to hypertension in ESRD patients. Although ultrafiltration effectively removes water and sodium concurrently through convection, there may be an additional exchange of sodium from the dialysate to the patient’s plasma depending on the sodium concentration gradient between the two compartments. Directly programmable ultrafiltration enables greater convective sodium removal in shorter periods of time, but increases the risk of intradialytic hypotension related to the abrupt hemodynamic changes. Although the use of higher dialysate sodium concentrations may reduce the risk of intradialytic hypotension, it may increase the risk of hypertension.49 In contrast, the use of individualized dialysate sodium concentrations (as opposed to standardized concentrations, which may exceed the pre-HD plasma sodium of the patient) have been associated with lower pre-HD systolic BP in the context of decreased thirst and interdialytic weight gain.50
The Renin-Angiotensin-Aldosterone System and Hypertension
The RAAS has local and systemic effects that control BP by altering renal sodium reabsorption and vascular resistance. Renin released from juxtaglomerular cells cleaves angiotensinogen to Ang I, which is ultimately converted to Ang II by the ACE. Ang II causes vasoconstriction upon binding to angiotensin type 1 (AT1) receptors in vascular smooth muscle cells (VSMCs). Ang II increases proximal tubular reabsorption of sodium and stimulates aldosterone release from the adrenal gland, which is responsible for further sodium reabsorption in the distal nephron. Although this proposed sequence applies to the levels and activity of RAAS components in the plasma, there is also local RAAS activity. Consequently, measurements of RAAS mediators in the plasma may not always identify a disruption of the axis at the tissue level.51
Early evidence for systemic RAAS activation in CKD stems from studies in patients with autosomal dominant polycystic kidney disease (ADPKD). It has been shown that PRA and plasma aldosterone were higher in hypertensive ADPKD patients compared to patients with essential hypertension, and plasma aldosterone is higher in normotensive ADPKD patients compared to healthy controls (despite similar creatinine clearance between the groups).52 Autosomal dominant polycystic kidney disease patients also manifest an accentuated response to ACE inhibitors compared to unaffected family members, further supporting the role of RAAS in the hypertension seen in ADPKD even prior to the onset of significant renal impairment.53 However, the findings from these studies may not be entirely generalizable to more heterogeneous groups of CKD patients because the proposed renal ischemia induced by large cyst formation in ADPKD does not necessarily apply to CKD from other etiologies. There is evidence that PRA and aldosterone are higher in hypertensive CKD patients compared to healthy controls, essential hypertension patients, or even normotensive CKD patients.54,55 However, there is also evidence that CKD patients have lower PRA compared to healthy
controls, although the values were similar to subjects with essential hypertension and normal renal function.56 Despite these conflicting findings, increased intrarenal RAAS activity has been described in patients with hypertension and varying etiologies of CKD, including immunoglobulin A (IgA) nephropathy, membranous nephropathy, and diabetic nephropathy.57,58,59
controls, although the values were similar to subjects with essential hypertension and normal renal function.56 Despite these conflicting findings, increased intrarenal RAAS activity has been described in patients with hypertension and varying etiologies of CKD, including immunoglobulin A (IgA) nephropathy, membranous nephropathy, and diabetic nephropathy.57,58,59
As mentioned previously, success in managing BP can be achieved in many ESRD patients by appropriately using ultrafiltration for volume removal.46 However, there are HD patients who remain quite hypertensive despite achieving their estimated dry weight. Previous reports have shown both increased or normal PRA in groups of HD patients compared to controls, and PRA did not consistently correlate with BP.60,61 However, increased PRA has been demonstrated in HD patients with hypertension that is ultrafiltration resistant compared to those whose BP responds to ultrafiltration.61 Bilateral nephrectomy, a procedure previously used for ultrafiltration-resistant hypertension in ESRD patients, has been shown to reduce PRA, Ang I and II, along with reductions in BP in these ultrafiltration-resistant hypertensive patients.62
Renin and Aldosterone
Although the primary action of angiotensin converting enzyme (ACE) inhibitors and ARB is to decrease the production and action of Ang II via the inhibition of the ACE enzyme or Ang II receptor, a complete perspective on the role of RAAS in hypertension associated with kidney disease warrants a discussion of the other components of the RAAS, including aldosterone and renin. Following the use of an ACE inhibitor or ARB, serum aldosterone levels typically decrease. However, in up to 40% of patients receiving these medications, aldosterone levels can rebound to pretreatment levels through a process referred to as aldosterone escape.63 Aldosterone increases BP via enhanced sodium reabsorption in the distal nephron, but it is also likely involved in vasoconstriction via interaction with the Ang II receptor.64 Despite evidence that CKD patients experiencing aldosterone escape may have worse control of proteinuria, systemic BP does not seem to be different from CKD patients whose aldosterone levels remain depressed following ACE inhibitor or ARB treatment.65,66 These studies included patients with IgA nephropathy who were normotensive and had creatinine clearance >50 mL per minute or patients with early diabetic nephropathy and hypertension; such investigations have not been performed in broader groups of CKD patients. Evidence regarding the potential benefit of mineralocorticoid receptor blockers is included in the Treatment section of this chapter.
In contrast to the expected decrease in aldosterone following ACE inhibitor or ARB use, renin levels and PRA are expected to increase as a result of the blockade in events downstream from the main actions of renin. It has been demonstrated in vivo that Ang II can be generated by enzymes other than ACE.67 This presents a potential obstacle to complete RAAS blockade if renin levels are sufficiently elevated in the context of an ACE inhibitor or ARB administration, and the possible use of add-on therapy to ACE inhibitors or ARB with a direct renin-inhibitor is further discussed in the Treatment section.
Angiotensin Converting Enzyme 2
Understanding of the RAAS continues to expand, and attention has been drawn to another enzyme in this pathway. ACE2 is a monocarboxypeptidase homolog of ACE that decreases Ang II levels by (1) increasing the degradation of Ang II to Ang 1-7, a vasodilatory and antiproliferative mediator, and (2) converting Ang I to Ang 1-9. This latter process not only prevents the conversion of Ang I to Ang II via ACE, but the increase in Ang 1-9 acts as a substrate for additional enzymatic conversion to Ang 1-7. Infusion of human recombinant ACE2 (rACE2) does not alter BP significantly in normotensive mice.68 However, rACE2 prevents BP increases induced by the infusion of Ang II when they are infused together. Consistent with the proposed mechanism of ACE2, Ang II levels were significantly lower and Ang 1-7 levels were higher in mice receiving Ang II plus rACE2 compared to Ang II alone. ACE2 is highly expressed in the kidney and is believed to play a role in the progression of CKD via local activity of the renal RAAS. Mice that have undergone 5/6 nephrectomy have significantly reduced renal ACE2 expression and a trend toward reduced renal ACE2 activity compared to sham-operated mice.69 Although BP was similar in nephrectomy and sham-operated mice, there was greater proteinuria in the nephrectomy mice. The proteinuria was further increased following administration of the ACE2 inhibitor. The major implications of these studies are that there may be further opportunities to reduce Ang II levels beyond the currently implemented strategies.
Sympathetic Nervous System and Hypertension
The SNS has been studied extensively as a possible contributor to hypertension in CKD and ESRD patients. In CKD patients, there is evidence for multiple pathways that the SNS may affect to increase BP. In support of the previous discussion of the effects of salt balance on BP, renal sympathetic nerve stimulation increases proximal tubular reabsorption of sodium and water.70 Additionally, the systemic effects of the SNS include increased cardiac output and vasoconstriction. Although evidence of activated SNS activity based on elevated catecholamine levels in CKD is inconsistent and possibly confounded by decreased renal clearance of these compounds, other estimates of SNS, such as muscle sympathetic nerve activity (MSNA), confirm the hyperactive state in CKD and ESRD. The mechanisms responsible for hyperactive SNS are thought to be related to increased renal nerve activity. The kidney possesses baroreceptors and chemoreceptors that increase renal nerve firing secondary to pressure changes or metabolites produced in response to ischemia
or uremia. Animal models of renal artery stenosis, arterial ligation causing partial renal ablation, or intrarenal phenol injection all reveal increased nerve activity.71,72,73 Activation of renal nerves results in a centrally mediated hypertension via activation of the SNS, which can ultimately be interrupted by a blockade of neural signals. For example, Sprague-Dawley rats that have undergone a 5/6 nephrectomy experience an attenuation in hypertension following a dorsal rhizotomy with a concurrent reduction in the turnover of norepinephrine in the hypothalamic nuclei and locus coeruleus.72 In humans, there is evidence for SNS activation even prior to overt renal impairment if a potential cause for renal ischemia is present. Hypertensive ADPKD patients have increased MSNA compared to controls despite preserved renal function in both groups.74 Muscle sympathetic nerve activity was even higher in ADPKD patients with decreased GFR, but was not different between normotensive ADPKD patients and controls. Given the experimental association with bilateral renal ischemia, the possibility exists that the increased MSNA is related to increases in RAAS activation. One study confirmed the presence of increased MSNA in CKD patients, and showed that the administration of the ACE inhibitor enalapril caused a reduction in MSNA (relative to the decrease in BP it caused), whereas amlodipine increased MSNA.75 In a small study including patients with various etiologies of nondiabetic CKD and matched control, MSNA changed similarly in the patients and controls with variability in volume status, but MSNA was persistently higher in the CKD patients.76
or uremia. Animal models of renal artery stenosis, arterial ligation causing partial renal ablation, or intrarenal phenol injection all reveal increased nerve activity.71,72,73 Activation of renal nerves results in a centrally mediated hypertension via activation of the SNS, which can ultimately be interrupted by a blockade of neural signals. For example, Sprague-Dawley rats that have undergone a 5/6 nephrectomy experience an attenuation in hypertension following a dorsal rhizotomy with a concurrent reduction in the turnover of norepinephrine in the hypothalamic nuclei and locus coeruleus.72 In humans, there is evidence for SNS activation even prior to overt renal impairment if a potential cause for renal ischemia is present. Hypertensive ADPKD patients have increased MSNA compared to controls despite preserved renal function in both groups.74 Muscle sympathetic nerve activity was even higher in ADPKD patients with decreased GFR, but was not different between normotensive ADPKD patients and controls. Given the experimental association with bilateral renal ischemia, the possibility exists that the increased MSNA is related to increases in RAAS activation. One study confirmed the presence of increased MSNA in CKD patients, and showed that the administration of the ACE inhibitor enalapril caused a reduction in MSNA (relative to the decrease in BP it caused), whereas amlodipine increased MSNA.75 In a small study including patients with various etiologies of nondiabetic CKD and matched control, MSNA changed similarly in the patients and controls with variability in volume status, but MSNA was persistently higher in the CKD patients.76
The evidence for the role of increased SNS activity in the etiology of hypertension in renal disease is best supported by the findings of Converse et al.77 Twenty years ago it was established that ESRD patients had increased MSNA compared to healthy controls and other ESRD patients that had undergone bilateral nephrectomies. Higher mean arterial BP was also seen in the ESRD patients whose native kidneys were still present, and the hypothesis was that afferent signaling from the ischemic kidneys modulated an overall SNS response best managed by surgical removal of the kidneys. This is further supported by the fact that reversing the uremic state of ESRD patients with renal transplantation does not decrease MSNA, but a native kidney nephrectomy in the transplant recipient does.78 The fact that nondiabetic HD patients have a normal arterial and cardiopulmonary baroreflex response weakens any suggestion that uremia-induced impairments in these reflexes might further contribute to abnormally elevated SNS activity in this population.79
Renalase
Adding to the evidence that CKD patients, particularly those with hypertension, have increased MSNA and circulating catecholamine levels is the recent discovery of a protein that may facilitate higher levels of catecholamines. Renalase is an amine oxidase that contributes to catecholamine degradation, which, in comparison to other common amine oxidases, can circulate in the plasma. Renalase is secreted into the circulation by the kidneys, and patients with ESRD on HD have nearly undetectable levels, supporting the role of adequate renal function to maintain its presence.80 Although in CKD patients, there is no direct evidence how circulating renalase is responsible for hypertension, animal models demonstrate the development of hypertension following knockout of the renalase gene and a reduction in BP following the infusion of recombinant renalase in Sprague-Dawley rats.80
ENDOTHELIAL CELL DYSFUNCTION AND HYPERTENSION
Because the increase in peripheral vascular resistance also contributes to the elevated BP in patients with kidney disease, it is important to understand the mechanisms responsible for vasoconstriction. Blood vessels are lined with endothelial cells, which release mediators that exert their actions on VSMC receptors. The balance between vasoconstricting mediators and vasodilating mediators dictates the ultimate response of the VSMC and the amount of resistance. Endothelial nitric oxide synthase (eNOS) uses arginine as a substrate to produce the vasodilator nitric oxide (NO). This process is dependent on the presence of the cofactor tetrahydrobiopterin (BH4), and production of NO can be inhibited by the arginine analog asymmetric dimethylarginine (ADMA). One of the primary vasoconstrictive agents is endothelin-1 (ET-1), but the ultimate response of the vascular tone is dependent on which ET receptor is being bound. The interplay between all of these mediators is complex. Substances such as Ang II modify the activity of ET-1, and NO release is sensitive to the relative state of oxidative stress and inflammation.
Endothelin
ET-1 is a 21 amino acid mitogenic peptide that is produced ubiquitously, but to a large extent in vascular endothelial cells. Its original description demonstrated that it had more potent vasoconstrictive effects than other vasoconstrictive peptides including Ang II, vasopressin, and neuropeptide Y while having a longer lasting effect on vascular tone than the endothelial-derived relaxing factor NO.81 Additionally, ET-1 has been shown to promote vascular cell hypertrophy and increase nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, resulting in oxidative stress and endothelial cell dysfunction.82 There are two primary receptors for ET-1: ET-A and ET-B. ET-1 binding to ET-A and ET-B receptors found on VSMC causes vasoconstriction, whereas binding to ET-B receptors on the endothelial cells causes vasodilation, suggesting a mechanism for feedback inhibition to stimulation by ET-1. Because ET-1 has paracrine behavior and migrates from the endothelial cells to VSMC (away from the lumen), plasma levels of ET-1 have proven to be unreliable in establishing a clear causal relationship with hypertension. However, mRNA expression of ET-1 in the endothelium of resistance vessels is higher in patients with moderate-to-severe hypertension compared to those with mild hypertension
or controls.83 Its role in hypertension in humans is further implicated by its effects on arterial tone. Following an infusion of ET-1, forearm blood flow decreases (increased tone) in both hypertensive and healthy humans, although the effect is greater in hypertensive patients. The pharmacologic inhibition of ET-A receptors alone or the combined inhibition of ET-A and ET-B receptors results in significant increases in forearm blood flow in the hypertensive patients, but not the healthy subjects.84 The administration of a nonselective ET receptor blocker in a dose of >500 mg per day resulted in reductions in systolic and diastolic office and ambulatory BP as compared to placebo; the BP reduction from the drug was similar to that achieved with the ACE inhibitor enalapril.85
or controls.83 Its role in hypertension in humans is further implicated by its effects on arterial tone. Following an infusion of ET-1, forearm blood flow decreases (increased tone) in both hypertensive and healthy humans, although the effect is greater in hypertensive patients. The pharmacologic inhibition of ET-A receptors alone or the combined inhibition of ET-A and ET-B receptors results in significant increases in forearm blood flow in the hypertensive patients, but not the healthy subjects.84 The administration of a nonselective ET receptor blocker in a dose of >500 mg per day resulted in reductions in systolic and diastolic office and ambulatory BP as compared to placebo; the BP reduction from the drug was similar to that achieved with the ACE inhibitor enalapril.85
In patients with CKD, both systemic and local renal effects are proposed to contribute to hypertension in this population. Partial nephrectomy Sprague-Dawley rat models suggest that there is an imbalance of ET expression and degradation in the uremic state. In these animals, there were increased ET-1 levels (related to increased expression) in the endothelial cells of the thoracic aorta and renal cortex.86 It was also found that the number of ET-B receptors was decreased in these locations, but there was a mild increase in ET-A receptor expression in the VSMC. Downregulation of ET-B receptors disables a mechanism for ET-1 degradation and can further contribute to the increased presence and action of ET-1.87,88
In CKD, there is also increased urinary and plasma levels of ET-1, independent of BP.89,90 The renal excretion rate of ET-1 is increased in hypertensive CKD patients, but not in subjects with normal renal function who have increased plasma ET-1 levels accompanying essential hypertension. These findings suggest that renal production of ET-1 increases as renal function declines and contributes to hypertension in CKD.91




Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
