Javier Fernández, Vicente Arroyo Hepatorenal syndrome (HRS) is a potentially reversible functional renal failure that occurs in patients with acute or chronic liver disease, advanced hepatic failure, and portal hypertension. Although it may occur in patients with acute liver failure or severe acute alcoholic hepatitis, it is mainly observed in patients with advanced cirrhosis. HRS is characterized by impaired renal function and marked abnormalities in the arterial circulation and endogenous vasoactive systems. In the kidney, there is pronounced vasoconstriction resulting in low glomerular filtration rate (GFR). In the splanchnic circulation, there is marked arteriolar vasodilation resulting in reduction of systemic vascular resistance and arterial hypotension.1–3 Two forms of HRS can be identified on the basis of the progression of the disease (Box 76-1). The acute form (type 1 HRS) is characterized by an acute and rapid deterioration in renal function that occurs in the setting of multiorgan failure (acute-on-chronic liver failure), whereas the chronic form (type 2) has an insidious onset and is characterized by moderate renal failure that follows a steady or slowly progressive course.1–3 Pseudohepatorenal syndrome describes concurrent hepatic and renal dysfunction secondary to a wide variety of infectious, systemic, circulatory, genetic, and other diseases and after exposure to a variety of drugs and toxins (Table 76-1).4 All these entities must be excluded before the diagnosis of HRS can be established. In all these conditions, the liver does not play a causative role in the pathogenesis of renal failure. Pseudohepatorenal syndrome is usually easy to exclude because the causative agent is frequently known and both renal and liver functional abnormalities are often found at first clinical presentation, when there is no evidence of advanced liver failure and portal hypertension. In contrast, HRS invariably occurs after liver failure and portal hypertension are fully established and frequently develops when the patient is undergoing treatment for these conditions or their complications. Table 76-1 Causes of pseudohepatorenal syndrome. * Accidental poisoning after ingestion of mushrooms of the Amanita genus. † Accidental poisoning after ingestion of the raw gallbladder or bile of the grass carp (a common practice in rural East Asia). ‡ Hemolysis, elevated liver enzymes, low platelet count. (Data from reference 10.) In HRS, reduction in GFR occurs mainly as a result of renal cortical hypoperfusion after intense cortical renal vasoconstriction, which can be demonstrated angiographically as marked beading and tortuosity of the interlobular and proximal arcuate arteries and the absence of a distinct cortical nephrogram and vascular filling of the cortical vessels (Fig. 76-1).5 Intense renal vasoconstriction is the final consequence of marked systemic circulatory dysfunction, characterized by progressive splanchnic arterial vasodilation, reduction of the effective arterial blood volume, hypotension, and homeostatic activation of the vasoconstrictor systems.3 This further compromises renal perfusion because intense renal vasoconstriction results in blunting of the autoregulation of renal blood flow, so that renal perfusion becomes more pressure dependent. In HRS, filtration fraction is also reduced, reflecting a dominant increase in afferent arteriolar tone and a decrease in the ultrafiltration coefficient. Serial systemic hemodynamic studies showed that HRS occurs in the setting of reduced mean arterial pressure (MAP), cardiac output, and wedge pulmonary pressure without change in systemic vascular resistance. These findings suggest that the inability of the heart to increase its output to compensate for a decrease in cardiac preload (secondary to the accentuation of splanchnic arterial vasodilation) also contributes to the pathogenesis of HRS.6 Vasoconstriction is not confined to the renal vascular bed. In HRS it is also observed in other extrasplanchnic territories, including the liver, brain, muscle, and skin.2,3 The renal and systemic hemodynamic changes that characterize HRS are a direct consequence of neurohumoral disturbances.2,3 Activation of the vasoconstrictor systems (the renin-angiotensin-aldosterone system [RAAS], the sympathetic nervous system, and vasopressin) is the cause of renal vasoconstriction; meanwhile, activation of the vasodilator systems occurs mainly in the splanchnic circulation and leads to splanchnic vasodilation. Increases in the serum and urinary levels of vasoconstrictors and in the plasma level of vasodilators are observed in patients with HRS. The vasoconstrictors include renin, norepinephrine, neuropeptide Y, arginine vasopressin, endothelin and F2 isoprostanes, and urinary cysteinyl leukotrienes; the vasodilators include plasma endotoxin, nitrite and nitrate (end product of nitric oxide metabolism), and glucagon. The sympathetic discharges through the renal nerves are also markedly increased. In contrast to increased plasma and urinary levels of vasoconstrictors and plasma level of vasodilators, decreased levels of urinary vasodilators have been observed in HRS. These include prostaglandin E2, 6-keto-prostaglandin F1 (a stable metabolite of renal prostacyclin), and kallikrein. Because the level of these urinary vasodilators is normal in compensated cirrhosis and higher than normal in decompensated cirrhosis with ascites and normal renal function, it is postulated that a reduction in the renal synthesis of renal vasodilators is the final event that leads to the development of HRS.3 Most of these neurohumoral abnormalities found in HRS are also detected, albeit to a lesser extent, in decompensated cirrhosis (with ascites) with normal renal function and in compensated cirrhosis (without ascites). These findings support the hypothesis that HRS most likely represents one end of the spectrum of homeostatic abnormalities that occur in liver failure and portal hypertension. Figure 76-2 shows the pathogenetic events that lead to HRS. Liver failure and portal hypertension through endotoxemia and increased shear stress increase vascular production of vasodilators, including nitric oxide, carbon monoxide, and glucagon in the splanchnic circulation, leading to the initiating event of splanchnic arteriolar vasodilation (the peripheral arterial vasodilation hypothesis). Splanchnic vasodilation leads to a decrease in systemic vascular resistance, but MAP is initially maintained by an increase in cardiac output, resulting in a hyperdynamic circulation. Splanchnic vasodilation also decreases arterial filling and reduces the effective arterial blood volume. The subsequent stimulation of the central volume baroreceptors leads to compensatory activation of the vasoconstrictor systems, in particular the arginine vasopressin system, RAAS, and sympathetic nervous system (including its hormones norepinephrine and neuropeptide Y), which help restore effective arterial blood volume. This restoration is achieved in patients with compensated cirrhosis but not in patients with decompensated cirrhosis, in whom progressive splanchnic arteriolar vasodilation leads to increased splanchnic capillary pressure, resulting in an increase in lymph formation that exceeds reabsorption capacity. In parallel, further contraction of the effective arterial blood volume leads to reduction of systemic MAP and further stimulation of the vasoconstrictor systems, resulting in sodium and water retention. The net result of these combined effects is continuous ascites formation (the forward theory of ascites formation).2,3 The splanchnic circulation is resistant to the effects of vasoconstrictors because of local release of vasodilators; progressive splanchnic vasodilation continues to occur as liver failure and portal hypertension progress. This leads to continued contraction of effective arterial blood volume, which, in combination with the progressive inability of the cirrhotic heart to respond to reduced preload consequent to splanchnic vasodilation, results in further reduction of MAP and more intense stimulation of the vasoconstrictor systems. Normally, the effect of vasoconstrictors on the renal circulation is counterbalanced by the reactive production of intrarenal vasodilators. It is postulated that HRS develops when the balance of activities between the renal vasoconstrictors and intrarenal vasodilators finally breaks down. The likelihood that this will occur increases with progressive or acute deterioration in liver function or increasing severity of portal hypertension (e.g., after acute alcoholic hepatitis) and is precipitated by events that lead to further volume contraction and reduction of the effective arterial blood volume (e.g., spontaneous bacterial peritonitis; see later discussion). The possibility for development of HRS in the cirrhotic patient is estimated to be 18% at 1 year and 39% at 5 years.7 Neither the cause nor the Child-Pugh score or the model for end-stage liver disease (MELD) score (http://www.mdcalc.com/child-pugh-score-for-cirrhosis-mortality/) predicts the incidence of HRS. Rather, independent predictors of HRS include dilutional hyponatremia, impairment in systemic hemodynamics (high plasma renin activity and noradrenaline concentration and low cardiac output),6,7 abnormal renal duplex Doppler ultrasound study findings (resistive index >0.7),8 and low GFR.3 Type 1 and type 2 HRS are considered to be different syndromes rather than different expressions of a common underlying disorder.2,3 Type 1 HRS is characterized by a rapid decline in renal function (see Box 76-1) and is observed in patients with acute decompensation of advanced cirrhosis, severe acute alcoholic hepatitis, or acute liver failure. In addition to developing a rapidly progressive acute kidney injury (AKI), patients also develop multiorgan dysfunction including severe hepatic failure (jaundice, coagulopathy), brain failure (hepatic encephalopathy), and frequently relative adrenal insufficiency. Hyponatremia is almost always present, and arterial blood pressure is usually low. Type 1 HRS may be precipitated by bacterial infections (especially spontaneous bacterial peritonitis), gastrointestinal bleeding, or total paracentesis without albumin administration. Left untreated, type 1 HRS tends to run a rapid and progressive downhill course, resulting in death of the patient within 2 to 3 weeks.7 Type 2 HRS is characterized by insidious onset and slowly progressive deterioration of renal function. This is most often observed in patients with cirrhosis and portal hypertension. These patients tend to be less severely jaundiced and mainly present with refractory ascites caused by poor response to diuretics. Low-normal arterial blood pressure, modest prolongation of prothrombin time, and moderate or marked hypoalbuminemia and hyponatremia are usually present. Type 2 HRS tends to run a slowly progressive downhill course over months, which most likely reflects the natural course of the disease because additional precipitating factors are not usually identified.2,3,7 Mean survival time after onset of type 2 HRS is 6 months. Hepatorenal syndrome is, by definition, a functional renal disorder, and the presence of significant glomerular and/or tubular disease excludes the diagnosis. However, glomerular abnormalities, including mesangial expansion, capillary wall thickening, mesangial and capillary wall electron-dense deposits, and immune deposits of C3 and IgA, IgM, and IgG, are frequently found in cirrhotic patients with normal renal function and minimal urinary abnormalities. The presence of such glomerular abnormalities in a cirrhotic patient, therefore, does not exclude the diagnosis of HRS. Protrusion of the proximal tubular epithelium into the Bowman space (glomerulotubular reflux) is not specific for HRS and is found in other conditions associated with profound renal ischemia and terminal hypotension. Although early autopsy studies demonstrated normal tubular morphology in patients who had died of HRS, detailed light and electron microscopic studies have documented proximal tubular lesions consistent with ischemic injury. However, these lesions do not explain the renal failure and reduction in GFR observed in patients with HRS.3 The diagnostic criteria for HRS were established by the International Ascites Club in 19961 and have recently been revised (Box 76-2).9 The main differences between the new and old criteria include the exclusion of creatinine clearance as a measure of renal function because of the difficulty in obtaining accurate urine collection data, the removal of ongoing bacterial infection as an exclusion criterion so that treatment of HRS is not delayed in these patients, the substitution of saline with albumin as the preferred fluid for plasma volume expansion, and the removal of the minor criteria because they are not thought to be essential. The diagnosis of HRS is mainly one of exclusion and should be suspected in any patient with acute or chronic liver disease with advanced liver failure and portal hypertension who develops progressive renal impairment. Serum creatinine concentration and blood urea nitrogen are poor markers of GFR in cirrhosis.3 Cirrhotic patients may have significant renal impairment despite normal serum creatinine or blood urea nitrogen concentration values because they are frequently malnourished, with reduced lean body mass, and often have a low urea generation rate because of liver failure and low protein intake. Severe hyperbilirubinemia, which is often present in patients with HRS, interferes with the Jaffe reaction for creatinine quantification and may cause falsely low results. Enzymatic creatinine assays are less susceptible to high bilirubin levels. In cases of uncertainty, GFR may be assessed with use of 125I-iothalamate or chromium 51–labeled ethylenediaminetetraacetic acid (51Cr-labeled EDTA). Studies involving a low number of patients suggest that serum cystatin C could be an accurate GFR marker in the cirrhotic population.10 However, the real usefulness of serum cystatin C in the assessment of GFR in cirrhosis needs to be confirmed. In patients with preexisting liver failure, portal hypertension, and renal failure, the use of nephrotoxic agents (e.g., nonsteroidal anti-inflammatory drugs [NSAIDs] and aminoglycosides) must be stopped, and conditions leading to renal failure must be excluded by careful history, physical examination, urine examination, and ultrasound study before the diagnosis of HRS can be considered. The absence of shock or gastrointestinal bleeding, and excess gastrointestinal, peritoneal, or renal fluid loss must also be documented. Prerenal AKI must be excluded by withdrawal of diuretics and fluid challenge either with 1.5 liters of normal saline or preferably with albumin, 1 g/kg body weight per day up to the maximum of 100 g/day for 2 days. Absence of microhematuria and proteinuria of less than 500 mg/day help exclude significant coexisting glomerular or tubulointerstitial disease leading to renal failure and support the diagnosis of HRS. The most important differential diagnosis for renal failure in cirrhosis is between type 1 HRS and “true” AKI with tubular damage, because they require rapid therapeutic decisions with different treatments. The parameters traditionally used to differentiate AKI with tubular damage from functional renal failure (urinary sodium excretion and urinary-plasma osmolality ratio) are of no value in patients with cirrhosis and ascites.3
Hepatorenal Syndrome
Definition
Pseudohepatorenal Syndrome
Causes of Pseudohepatorenal Syndrome
Potential Causes
Predominantly Tubulointerstitial Involvement
Predominantly Glomerular Involvement
Infections
Sepsis, leptospirosis, brucellosis, tuberculosis, Epstein-Barr virus, hepatitis A virus
Hepatitis B and C viruses, HIV infection, Schistosoma mansoni, liver abscess
Drugs
Tetracycline, rifampin, sulfonamide, phenytoin, allopurinol, fluroxene, methotrexate (high dose), acetaminophen overdose
Toxins
Carbon tetrachloride, trichloroethylene, chloroform, elemental phosphorus, arsenic, copper, chromium, barium, amatoxins,* raw carp bile toxins†
Systemic diseases
Sarcoidosis, Sjögren syndrome
Systemic lupus erythematosus, vasculitis, cryoglobulinemia, amyloidosis
Circulatory failure
Hypovolemic or cardiogenic shock
Malignancy
Lymphoma, leukemia
Congenital and genetic disorders
Polycystic liver and kidney disease, nephronophthisis, congenital hepatic fibrosis
Miscellaneous
Fatty liver of pregnancy, Reye syndrome
Eclampsia, HELLP syndrome,‡ cirrhotic glomerulopathy
Pathophysiology and Pathogenesis
Circulatory Dysfunction: Renal and Systemic Hemodynamic Changes
Neurohumoral Abnormalities
Summary of Pathogenetic Events
Epidemiology
Clinical Manifestations
Pathology
Diagnosis and Differential Diagnosis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Hepatorenal Syndrome
Chapter 76
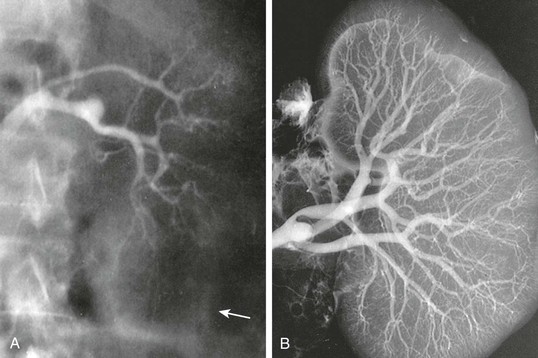
Figure 76-1 Renal arteriography of a patient with hepatorenal syndrome (HRS). A, Renal angiogram (the arrow marks the edge of the kidney). B, The angiogram carried out in the same kidney at autopsy. Note complete filling of the renal arterial system throughout the vascular bed to the periphery of the cortex. The vascular attenuation and tortuosity seen previously (A) are no longer present. The vessels are also histologically normal. This indicates the functional nature of the vascular abnormality in HRS.4
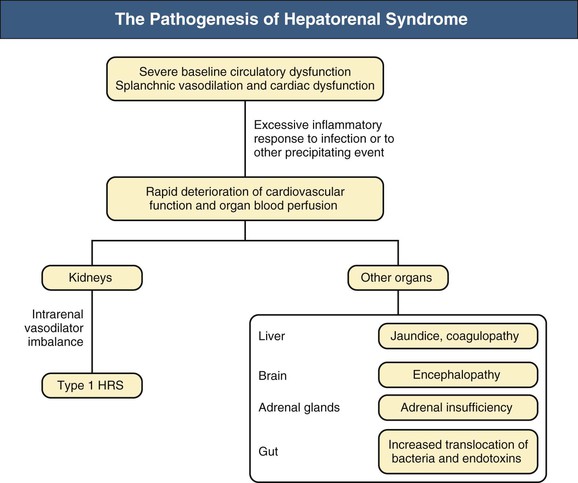
Figure 76-2 The pathogenesis of hepatorenal syndrome (HRS). Mechanisms leading to type 1 HRS and multiorgan failure. Patients with cirrhosis and ascites present a severe cardiocirculatory dysfunction that may be further aggravated by infection. Although circulatory dysfunction predominantly affects the kidneys and leads to the development of type 1 HRS, it also decreases the perfusion of other organs and systems such as the liver, with marked impairment in hepatic function and aggravation of portal hypertension; the brain, with the development of hepatic encephalopathy; the adrenal glands, with the development of relative adrenal dysfunction; and the gut, decreasing intestinal motility and promoting intestinal bacterial overgrowth and bacterial translocation.2,3






