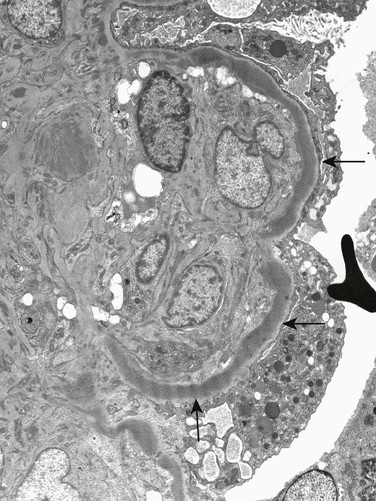
H. Terence Cook, Matthew C. Pickering Glomerular diseases associated with abnormalities of the complement system include thrombotic microangiopathy and glomerulonephritides. Activation of complement in most cases of glomerulonephritis (GN) is secondary to other processes in the glomerulus, such as deposition of immune complexes. In a small number of cases, however, abnormalities of the complement system itself, both genetic and acquired, are the cause of GN, and these patients are the focus of this chapter. Abnormalities of the classical pathway of complement have been associated with GN. Most important, deficiencies in early components of the classical pathway of complement have been associated with autoimmunity and GN.1 This is most clearly seen in the rare individual with C1q deficiency, who typically has a lupus-like illness. This is thought to result from failure of clearance of immunogenic apoptotic bodies and other cellular debris in the absence of normal classical pathway complement activation. The largest group of patients with complement abnormalities and GN has an abnormality of control of the alternative pathway of complement activation, with glomerular deposition of C3 in the absence of immunoglobulin, now termed C3 glomerulopathy (Box 22-1). A recently introduced term, C3 glomerulopathy encompasses glomerular disease characterized by the accumulation of complement component C3 in glomeruli caused by abnormal control of complement activation, deposition, or degradation, particularly abnormal control of the alternative pathway of complement activation.2 Characteristically, glomeruli show strong immunohistologic staining for C3 without significant staining for immunoglobulins or for components of the classical pathway of complement activation, C1q and C4. Thus defined, C3 glomerulopathy is distinct from atypical hemolytic uremic syndrome (HUS), which may also be associated with alternative pathway activation; in HUS, complement activation is on the renal endothelium and on electron microscopy (EM) is not associated with well-defined deposits (see Chapter 29). C3 glomerulopathy may show a variety of appearances on light microscopy, including mesangial proliferation, a membranoproliferative pattern, endocapillary proliferation, and crescent formation. It is now recognized that many cases previously classified morphologically as “membranoproliferative glomerulonephritis” (MPGN) are cases of C3 glomerulopathy. This includes cases that had been classified as MPGN types I, II, or III. Indeed, it appears that most cases previously called MPGN type III are examples of C3 glomerulopathy (see Chapter 21). On EM, C3 glomerulopathy may have a variable appearance as well. However, a common presentation is of dense deposit disease (DDD). This condition is characterized by replacement of the basement membrane by dense bands on EM (Fig. 22-1). In some cases, the light microscopy appearance in DDD resembles membranoproliferative glomerulonephritis, which was previously designated “MPGN type II.” However, most cases of DDD do not have MPGN morphology on light microscopy. Cases of C3 glomerulopathy that do not have typical highly dense deposits of DDD show a range of appearances on EM, with deposits that may be mesangial, subendothelial, or subepithelial, and that may be more or less well defined. These cases of non-DDD C3 glomerulopathy have been given the collective name of C3 glomerulonephritis (C3GN).3,4 The pathogenesis of C3 glomerulopathy involves a dysregulation of the alternative pathway of complement (see Fig. 29-10). In health, the alternative pathway is constantly being activated, but at an extremely low rate. With constant generation of small amounts of activated C3, this low-grade activation allows the pathway to be rapidly switched on when needed. In the presence of pathogens, rapid amplification of C3b is achieved through a positive feedback loop termed the C3b amplification loop that can generate millions of C3b molecules within minutes. Because this amplification can progress so rapidly, efficient systems are required to prevent inappropriate activation of the pathway. The most important protein in the circulation that controls the alternative pathway is complement factor H (CFH). CFH exerts this control in three ways: (1) blocking the formation of alternative pathway C3 convertases by binding to C3b and thus inhibiting interaction between C3b and factor B, (2) promoting the spontaneous dissociation of these convertases, and (3) working with another plasma protein, factor I, to cleave C3b to iC3b. Mice that have been genetically engineered to lack factor H have undetectable circulating C3 because their C3 is constantly consumed by the uncontrolled alternative pathway.5 Factor H is an abundant single-chain glycoprotein predominantly made in the liver. It is composed of protein subunits, termed short consensus repeat (SCR) domains. The activity of CFH can be modulated by a group of closely related proteins called factor H–related proteins (CFHRs), five of which exist in humans. CFHRs are coded for by genes adjacent to the gene for CFH and have a similar structure to CFH. The high degree of homology of these genes has led to a number of recombination and deletion events that have resulted in both common polymorphisms and rare pathogenic mutations at this locus. The most common polymorphism is a deletion of CFHR1 and CFHR3 that is present in homozygosity in 5% to 20% of healthy individuals, depending on ethnic origin. It is now clear that the CFHRs are able to compete with the binding of CFH to C3b in some circumstances.6 CFHRs, unlike CFH, are unable to inhibit complement activation and thus are now thought to antagonize the action of CFH. In many cases of C3 glomerulopathy, the pathogenesis is a failure of CFH to control the activation of the alternative pathway in the circulation, which is associated with low levels of circulating C3 because of uncontrolled consumption. Up to 80% of patients with DDD and up to half of patients with C3GN have low levels of serum C3.3 Many of these patients have a C3 nephritic factor (C3NeF). C3NeFs are autoantibodies that are able to stabilize the alternative pathway C3 convertase by preventing CFH from carrying out its normal functions. It therefore seems likely that C3NeF plays an important etiologic role in these patients. However, the role of C3NeF is complicated because C3NeFs may also be found in patients with other forms of GNs and even in healthy patients. In other cases, genetic mutations lead to failure of alternative pathway control, including patients with complete CFH deficiency caused by gene deletion, mutations in CFH that interfere with its binding to C3b, and mutations in C3 that change its structure to prevent its inhibition by CFH. In some patients, failure of alternative pathway control is associated with autoantibodies directed against factor H that target its regulatory domain. In cases of C3 glomerulopathy not associated with excessive activation of C3 in the circulation, the nephrologist can assume that there is a failure to control the alternative pathway locally within the glomerulus. This might be caused by a failure to control activation or by inappropriate handling of the fragments of C3 generated by alternative pathway activation. In most cases the pathogenesis is still not clear, but there is an example of a familial form of C3 glomerulopathy, called CFHR5 nephropathy, without systemic C3 activation. This is a common cause of kidney disease in the population of Cyprus, in which the mutation is a duplication of the first two exons of the CFHR5 gene.6 This leads to an abnormal protein that forms multimers able to deregulate the activity of CFH on surfaces.7 Therefore, the abnormal protein interferes with the action of CFH locally within the glomerulus and enhances alternative pathway activation.
Glomerulonephritis Associated with Complement Disorders
Definitions
C3 Glomerulopathy
Etiology and Pathogenesis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Glomerulonephritis Associated with Complement Disorders
Chapter 22