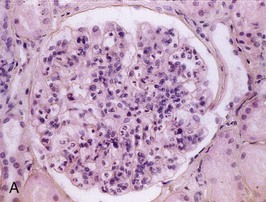
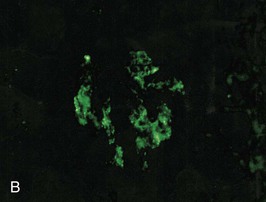
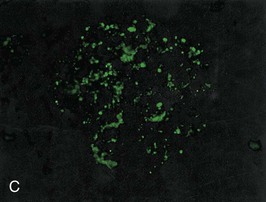
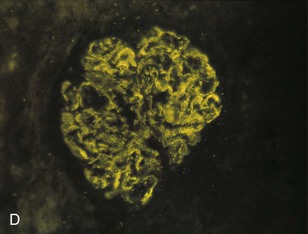
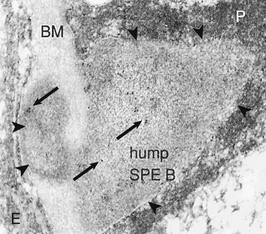
Bernardo Rodriguez-Iturbe, Emmanuel A. Burdmann, Rashad S. Barsoum The observation of dark scanty urine in the convalescent period of scarlet fever is more than two centuries old. At the beginning of the last century, Clemens von Pirquet, on the basis of clinical observations, postulated that the disease resulted from antibodies that instead of having beneficial effects were pathogenic—an insight that opened the field of immune-mediated renal disease. Today, the incidence of infection-related glomerulonephritis (GN) is declining in developed countries, and usually the disease is associated with older individuals and debilitating conditions such as diabetes, malignant neoplasia, acquired immunodeficiency syndrome (AIDS), and alcoholism. The prognosis in elderly individuals is poor, with less than 25% recovering full renal function. In recent years there has been a tendency for an increase in GN associated with Staphylococcus (IgA dominant) and fewer cases associated with Streptococcus, but a wide variety of pathogens have been reported (Box 57-1). In general, the infection-related GNs have a better prognosis than other forms of GN, but in adult patients with other comorbid factors they carry a significantly higher risk of chronic kidney disease (CKD).1 The histologic patterns and the most typical infectious diseases associated with them are shown in Table 57-1. Mesangial proliferative GN is usually acute and self-limited. Microscopic hematuria and non-nephrotic proteinuria are present in association with deposits of IgG, IgM, and C3. Serum complement may be transiently decreased. Diffuse proliferative GN is also acute and self-limited if the infection is eradicated. IgG, IgM, and C3 deposits are prominent in the mesangium and glomerular capillaries, and electron-dense deposits are present in mesangial, subendothelial, and subepithelial locations. Staphylococcal infections, particularly methicillin resistant, may induce diffuse proliferative GN with crescent formation and exclusive or predominant IgA deposition. Membranoproliferative GN (MPGN) is commonly observed with chronic infections. The clinical presentation is usually with severe proteinuria, microscopic hematuria, and variable degrees of hypertension. Immunoglobulins and C3 deposits are present. In the setting of liver disease (as in Schistosoma mansoni infection), IgA may be a predominant component of the immune deposits. Membranous nephropathy (MN) is associated with chronic infections and may manifest as the nephrotic syndrome. Finally, vasculitis may develop in association with viral (especially hepatitis B virus [HBV] and human immunodeficiency virus [HIV]) or bacterial (rarely Streptococcus) infections. Infection-related GN, especially when it is immune complex associated, occurs more frequently and has a worse prognosis in conditions in which there is difficulty clearing the infection or immune complexes. These conditions include HIV infection, infections acquired in the neonatal period (when tolerance is often induced, such as is observed with HBV infection), chronic liver disease, diabetes, and chronic alcoholism. Glomerulonephritis associated with infections usually results from glomerular deposition of immune complexes containing bacterial antigens, but other mechanisms may also occur. Staphylococcal infections may cause GN induced by bacterial wall superantigens that induce a nonspecific polyclonal immunoglobulin response. Focal segmental glomerulosclerosis (FSGS) with glomerular collapse (collapsing glomerulopathy) may occur with HIV or parvovirus B19 infection. The hemolytic uremic syndrome (HUS) may result from verotoxin-producing Escherichia coli or Shigella species (see Chapter 29). Amyloidosis may result from chronic infections such as tuberculosis, leprosy, and schistosomiasis. Interstitial nephritis manifested as acute renal failure may result from several viral (especially Epstein-Barr virus [EBV]) or bacterial (especially Legionella) infections. Hantavirus can also induce a hemorrhagic fever–renal failure syndrome in which infection of the interstitial capillaries and tubules leads to acute renal failure (see Chapters 69 and 70). Poststreptococcal glomerulonephritis (PSGN) is more common in males (2 : 1) and usually affects children 2 to 14 years old. Certain strains of group A Streptococcus pyogenes are nephritogenic: impetigo caused by streptococci of M types 47, 49, 55, and 57 and upper respiratory infections with types 1, 2, 4, and 12.2 More recently, ingestion of unpasteurized milk contaminated with group C Streptococcus (Streptococcus zooepidemicus) has caused clusters of cases and at least one large epidemic.3 The risk of nephritis in epidemics may range from 5% in throat infections to as high as 25% in M type 49 pyoderma. The risk of PSGN is reduced by early antibiotic treatment. A genetic predisposition is suggested because there is an association of PSGN with HLA-DR4 and HLA-DR1 and higher attack rates in siblings than expected for the general population. Poststreptococcal glomerulonephritis is becoming less common in industrialized countries and, instead of affecting predominantly children, now occurs more commonly in debilitated elderly individuals, alcoholics, diabetics, and intravenous drug users. The reduction in incidence of PSGN likely relates to improvement in public health, more rapid and frequent use of antibiotics, and perhaps the common practice of fluorination of water, which reduces the expression of virulence factors in cultures of S. pyogenes. Nevertheless, PSGN remains common in developing countries, where it may have an incidence of 9.3 to 9.8 cases per 100,000 population,4 and it represents a major problem in indigenous populations. For example, in Australia more than 95% of the cases occur in aboriginal tribes in remote locations.5 Two nephritogenic streptococcal antigens have been identified: nephritis-associated plasmin receptor (NAPLr), which was characterized as glyceraldehyde-3-phosphate dehydrogenase (GAPDH); and streptococcal pyrogenic exotoxin B (SPEB) and its more immunogenic precursor, zymogen. GAPDH and SPEB have been demonstrated in renal biopsy specimens of acute PSGN, and antibody titers to both these antigens are elevated in most convalescent sera.2 GAPDH has been localized in areas of the glomeruli with plasmin-like activity, suggesting a local direct mechanism of glomerular inflammatory damage, but it is not co-localized with complement or immunoglobulin.6,7 In contrast, SPEB is co-localized with both complement and IgG, suggesting a participation in the immune-mediated glomerular damage.8 SPEB is the only putative streptococcal nephritogenic antigen that so far has been demonstrated in the subepithelial electron-dense deposits known as humps, the most typical histologic lesion of acute PSGN (Fig. 57-1). In studies from Latin America and central Europe, SPEB but not GAPDH is usually detectable in renal biopsy specimens of PSGN patients.9 In contrast, GAPDH was present in PSGN in a study of Japanese patients.6,7 These contrasting results raise the possibility that different streptococcal antigens are capable of inducing acute nephritis in different ethnic groups. Indeed, a study of the group C S. zooepidemicus strain that caused the epidemic outbreak in Brazil revealed an absence of the gene related to SPEB, which documents that this antigen was not involved in that epidemic.9 Poststreptococcal glomerulonephritis is thought to occur when persistent streptococcal infection results in antigenemia and the development of circulating immune complexes that primarily deposit in the subendothelial and mesangial locations to initiate an inflammatory cascade with local complement activation and the recruitment of neutrophils and monocytes-macrophages. Subepithelial immune deposits (humps) develop when cationic antigens (e.g., SPEB) are involved and result from dissociation of immune complexes in the subendothelial space with transit and re-formation on the outer aspect of the glomerular basement membrane. Several issues remain unresolved. For example, immune complex disease generally results in activation of the classical complement pathway, yet in most cases, C4 levels are normal and only C3 is found in the deposits. This could be explained by the presence of antigens (such as GAPDH) that activate the alternative pathway. In some patients, C3Nef IgG antibodies that are capable of activating the alternative complement pathway have been demonstrated in sera. Activation of the mannose-binding lectin complement pathway by bacterial antigens has been postulated (see Chapter 16), but individuals genetically unable to activate this pathway may still develop PSGN. Finally, the role of autoimmune mechanisms remains to be clarified. Rheumatoid factors (especially IgG rheumatoid factor) and cryoglobulins are found in the serum of one third of patients in the first week of the disease. Rheumatoid factors (antibody to IgG) have been shown in one third of the renal biopsy specimens and in the eluate from the kidney in a fatal case. Anti-IgG reactivity may result from the loss of sialic acid from autologous IgG due to streptococcal neuraminidase (sialidase) or from binding of the Fc fragment of IgG to type II Fc receptors in the streptococcal wall.2 Anti-IgG antibody, induced by streptococcal IgG Fc-binding protein, may play a role in the renal accumulation of IgG-containing complexes.10 Additional manifestations of autoimmune reactivity include the anti-C1q antibodies, particularly in severe cases, and rarely anti-DNA reactivity, antineutrophil cytoplasmic antibody (ANCA), and autoimmune hemolytic anemia.2 Renal biopsy shows a diffuse endocapillary GN with proliferation of mesangial and endothelial cells (see Fig. 57-1). There is glomerular and interstitial infiltration of monocytes and lymphocytes. Glomerular accumulation of neutrophils is common and is termed exudative GN. Glomerular immune deposits of C3 (100% of the cases), IgG (62%), IgM (76%), and properdin and the terminal membrane attack complex C5b-C9 (85% of the cases, usually co-deposited with C3) are seen in glomerular capillary loops and in the mesangium. A seminal work by Sorger and colleagues11 described three patterns of immunofluorescence in the glomeruli and their clinical correlations: the mesangial pattern of irregular and heavy immune deposits, the starry sky pattern of deposits scattered in mesangium and in capillary walls, and the garland pattern formed by gross deposits in the capillary loops (see Fig. 57-1). The garland pattern is clinically relevant because it is associated with heavy proteinuria and a large number of electron-dense subepithelial immune deposits. Ultrastructural studies demonstrate the subepithelial humps, which are typical, although not pathognomonic of PSGN because they may also be observed in postinfectious GN of other causes (classically endocarditis-associated GN secondary to Staphylococcus infection), cryoglobulinemia, and lupus nephritis. Recent reports have called attention to the identification of NAPLr antigen in biopsies with features of MPGN, raising the possibility of a more varied histologic presentation on PSGN than previously recognized. Glomerulonephritis resolves by apoptosis of the excess cells. Residual renal injury is common, and biopsy specimens obtained years later show variable degrees of focal glomerulosclerosis and mesangial expansion even in the absence of clinical disease. The prognostic significance of these changes is undetermined. Most patients give a history of a previous streptococcal infection, although it has often resolved at presentation. The incubation period is longer after skin infections (several weeks) than after throat infections (2 weeks). The classic presentation is acute nephritic syndrome. Hypertension is found in 80% of patients. Edema occurs in 80% to 90% of cases and is the chief complaint in 60%; yet ascites is distinctly unusual. Primary sodium retention is the cause of expanded intravascular volume, hypertension, and edema. Plasma renin activity and aldosterone are reduced and natriuretic peptide levels are increased. Hematuria is universal and in 30% of cases is macroscopic. The nephrotic syndrome may occur at the onset in 2% of the children and 20% of adults. Rapidly progressive renal failure, resulting from extracapillary crescent formation, occurs in less than 1% of patients. Increments in serum creatinine occur in 25% to 40% of the children and in up to 83% of adults. Positive cultures for Streptococcus are obtained in 10% to 70% of the cases during epidemics and in about 20% to 25% of sporadic cases. Antistreptolysin O (ASO) titers are increased in more than two thirds of patients with PSGN after throat infections, and anti-DNAse B titers are elevated in 73% of the post-impetigo cases. The streptozyme panel (which measures antibodies to four antigens, anti-DNAse B, antihyaluronidase, ASO, and antistreptokinase) is more sensitive, and the result is positive in more than 80% of patients. Antibody titers to GAPDH and SPEB/zymogen, although more sensitive and specific,2,4 are not clinically available. Serum C3 levels are depressed in more than 90% of patients in the first week of disease and return to normal in less than 2 months. Levels of C4, a measure of classical complement pathway activation, may be normal. Serum IgG and IgM are elevated in 80% of the cases, and in contrast with another poststreptococcal disease, rheumatic fever, IgA is normal. Cryoglobulins, elevated rheumatoid factor, and anti-C1q antibodies are present in up to one third of patients in the acute phase. Subclinical disease, manifested by microscopic hematuria and fall in serum complement, occurs four to five times more frequently than clinically overt disease and often involves siblings of index cases. On occasion, whole families have various manifestations of PSGN; it is therefore important to inquire about a history of streptococcal infections and signs of nephritic syndrome among family members. Renal biopsy is not routinely indicated in PSGN but may be required to confirm the diagnosis in the presence of unusual clinical features, such as nephrotic proteinuria, decreased C3 levels lasting for more than a month (suggests lupus or hypocomplementemic MPGN), or increasing renal dysfunction (suggests crescentic GN). Management includes culture and treatment of any remaining streptococcal infection. Early antibiotic treatment is likely to prevent PSGN. Treatment is with penicillin IM (a single intramuscular [IM] injection of 1.2 million units of benzathine penicillin in adults or half this dose in small children) or oral penicillin, or erythromycin (in patients allergic to penicillin) or cephalosporins. Oral treatments should be given in doses every 6 hours for 7 to 10 days.12 Treatment of the acute nephritic syndrome includes restriction of fluid and sodium intake and the use of loop diuretics to treat circulatory congestion. An oral long-acting calcium antagonist is usually sufficient to control hypertension. Nitroprusside is used in exceptional cases with hypertensive encephalopathy. Dialysis (either hemodialysis or peritoneal dialysis) is required in 25% to 30% of adults but seldom in children. Rare complications of acute PSGN include cerebral vasculitis, cerebral vasogenic edema, and posterior reversible leukoencephalopathy. The last is manifested by mental disturbances, visual hallucinations, headache, and convulsions and may be confused with hypertensive encephalopathy. The diagnosis requires the use of magnetic resonance imaging studies. Anecdotal reports suggest that the rare patients with crescentic GN associated with PSGN may benefit from pulse methylprednisolone therapy, and if spontaneous improvement is not observed in 2 weeks, this therapy may be tried. The prognosis of crescentic PSGN is significantly better than that of crescentic GN from other causes, but complete recovery may be expected in less than half the cases. Most children with PSGN recover. In older patients, there are higher acute complication rates including a higher prevalence of renal impairment (60% to 70%), congestive heart failure (40%), nephrotic proteinuria (20%), and mortality (25%).4 Deficiency of complement regulatory proteins (such as factor H–related protein 5) may represent a risk factor for the development of CKD in these patients.13 After recovery, mild proteinuria (<500 mg/day) and microscopic hematuria may persist for up to 1 year without worsening the long-term prognosis. Nevertheless, some individuals, especially adults, may have persistent impaired renal function, proteinuria, or hypertension. However, end-stage renal disease (ESRD) occurs in less than 1% of the children observed for one to two decades after the acute attack.4 Certain epidemics have reported a high incidence of chronicity, perhaps because of a predominantly adult population.14 Risk factors for the development of CKD include an onset with nephrotic-range proteinuria, older age, and coexistent diabetic nephropathy and metabolic syndrome.15 The epidemiology of infective endocarditis has changed in the last three to four decades, especially in industrialized countries. Instead of being a disease that affects predominantly young adults with rheumatic heart disease, it now is seen in older patients and in patients at risk, a category that includes drug users, patients with prosthetic valves and implantable devices, patients with HIV infection, and patients who develop health care–associated bacteremia. Staphylococcus aureus and Staphylococcus epidermidis are the most common pathogens in hospital-acquired infective endocarditis, whereas Streptococcus infections are more frequent in community-acquired and native valve endocarditis.16 Nevertheless, the incidence of methicillin-resistant S. aureus (MRSA) causing community-acquired endocarditis is increasing.17 Gram-negative bacteria (Enterococcus faecalis, E. coli, Brucella, and Proteus) less frequently cause infective endocarditis. Culture-negative infective endocarditis is usually caused by Bartonella species and Tropheryma whipplei.18 In a survey of 62 patients with infective endocarditis in whom renal tissue was available for study, biopsy and autopsy material demonstrated GN in 26%; localized infarcts, half of which were septic, in 31%; and interstitial nephritis, mostly attributable to antibiotics, in 10%. Cortical necrosis was found in 10% of the patients.19 The most common GN was diffuse proliferative, followed less frequently by focal GN, MN, and MPGN type I. Crescent formation is found in about half of the proliferative GN forms. Widespread deposition of IgM, IgG, and C3 and electron-dense subendothelial, mesangial, and subepithelial deposits (which resemble poststreptococcal humps) are usually evident. In subacute endocarditis, focal segmental proliferative lesions with fibrinoid necrosis or capillary thrombi and mesangial immune deposits may be present. Variable degrees of cellular infiltration and atrophy and fibrosis may be seen in tubulointerstitial regions. Intense eosinophilic infiltration should suggest another diagnosis, such as acute interstitial nephritis secondary to antibiotics.19 The pathogenesis of endocarditis-associated GN involves the deposition of immune complexes containing bacterial antigens in the glomeruli, a mechanism similar to that proposed for PSGN. Cryoglobulins (polyclonal, i.e., type III) are present in 50% of patients and may be found in glomeruli. Some bacteria (classically MRSA) express superantigens that can also activate T cells directly and lead to a polyclonal gammopathy and immune complex GN (see later). Patients often have fever, arthralgias, anemia, and purpura. Rarely, infective endocarditis may manifest as primary renal disease without characteristic systemic symptoms. Classic findings in endocarditis, such as Osler nodes, Janeway lesions, and splinter hemorrhages, are seldom seen, and the diagnosis may be missed until autopsy in 38% of the patients.20 The renal manifestations usually are microscopic hematuria and mild proteinuria (except in patients with membranous GN, which may have proteinuria in the nephrotic range), with or without mild impairment in renal function (except in patients with renal vasculitis, in which renal failure is common). A rapidly progressive clinical course or nephrotic syndrome is unusual. Decreased C3 and C4 levels (consistent with activation of the classical complement pathway), high titers of rheumatoid factor, circulating immune complexes, and type III cryoglobulins are common findings. These serologic findings are observed in 50% of patients with infective endocarditis and in a higher proportion of patients with endocarditis-associated GN, although complement levels may be normal in superantigen-mediated GN. The differential diagnosis includes antibiotic-associated nephrotoxicity, interstitial nephritis, and embolism. Embolism may originate from the left side of the heart or from the right if the foramen ovale is patent. Microscopic or large emboli may occlude small or large vessels, the latter observed with fungal or Haemophilus endocarditis. Large emboli may produce flank pain, hematuria, and pyuria; microemboli produce local infarcts and microabscesses that give the kidney the classic “flea-bitten” appearance. Interstitial nephritis is often associated with fever, eosinophilia, and eosinophiluria (see Chapter 62). Unfortunately, endocarditis-associated GN may also cause fever, and eosinophiluria can complicate crescentic nephritis of any etiology.
Glomerular Diseases Associated with Infection
General Characteristics of Glomerular Diseases Associated with Infection
Histologic Patterns and Pathogenesis
Bacterial Infections
Poststreptococcal Glomerulonephritis
Epidemiology
Pathogenesis
Pathology
Clinical Manifestations
Management
Prognosis
Endocarditis-associated Glomerulonephritis
Clinical Manifestations and Diagnosis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Glomerular Diseases Associated with Infection
Chapter 57
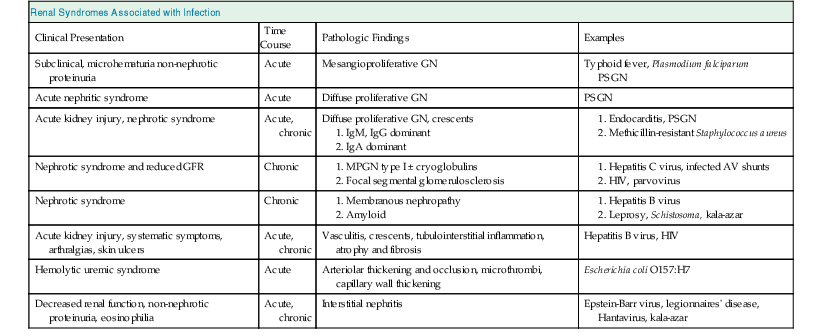
Figure 57-1 Poststreptococcal glomerulonephritis (GN). A, A diffuse proliferative and exudative GN can be seen with light microscopy. B to D, Immunofluorescence showing the mesangial (B), starry sky (C), and garland (D) patterns. E, Immune electron microscopy showing the characteristic subepithelial electron-dense deposits (humps) (arrowheads), inside which streptococcal pyrogenic exotoxin B (SPE B) is demonstrated (arrows) (immunogold staining). BM, Basement membrane; P, podocyte. (Reprinted with permission from reference 8.)






