Essentials of Diagnosis
- Focal segmental glomerulosclerosis (FSGS) can be primary or secondary.
- Diagnosis requires the presence of the characteristic histopathologic lesion.
- Genetic abnormalities in podocyte proteins may account for 25% of primary FSGS.
- The presenting complaint is usually proteinuria or nephrotic syndrome.
- Nearly 50% of cases progress to end-stage renal disease (ESRD) over 5–10 years and disease recurs in 30% of those who receive a kidney transplant.
- Failure to respond to corticosteroid treatment is a poor prognostic sign and there is no proven therapy in these patients.
General Considerations
FSGS is an important glomerulopathy because it has a high risk of progression to ESRD. It is not a distinct disease but rather represents a pattern of response to injury that probably originates in the podocyte. FSGS occurs in all ethnic groups, both genders, and all geographic locales. Recent data indicate that the incidence of FSGS is rising, especially in black patients. This has been confirmed in reviews of renal biopsy findings in the United States and Canada that demonstrate a 2- to 3-fold increase in the incidence of FSGS over the period from 1984 to 2002. In addition, according to the North American Pediatric Renal Transplant Collaborative Study, FSGS is the most frequent form of acquired renal disease necessitating renal replacement therapy in pediatric patients. Similarly, it is the most common cause of idiopathic nephrotic syndrome in adults and is a major cause of ESRD.
FSGS usually presents with asymptomatic proteinuria or overt nephrotic syndrome. In those patients who are diagnosed with isolated proteinuria, the abnormality is usually detected on a routine urinalysis. The clinical picture in those who present with nephrotic syndrome is almost indistinguishable from those with minimal change nephrotic syndrome (MCNS). Hematuria, evidence of tubular dysfunction such as glycosuria, hypertension, and mild azotemia, may be present in 15–30% of patients with new-onset nephrotic syndrome and these features may increase the clinical suspicion that a patient has FSGS. However, the key feature that prompts further investigation to establish the diagnosis of FSGS is failure to respond to a standard course of corticosteroids. It is this clinical finding that triggers the performance of a diagnostic renal biopsy; however, the utilization and timing of this procedure may differ among those who care for children or adults. Although the prognosis may be better in patients who present with subnephrotic-range proteinuria versus nephrotic syndrome, this difference has not been confirmed in patients of all ages.
The diagnosis of FSGS requires histopathologic evidence of segmental glomerular sclerosis and hyalinosis. The lesion often manifests in juxtamedullary nephrons during the early stages of disease and it can be associated with periglomerular scarring, tubular atrophy, and interstitial fibrosis in the vicinity of the affected glomerulus (Figure 25–1). Generally, immunofluorescence studies are unrevealing. Electron microscopy demonstrates foot process effacement, the absence of immune deposits, and mesangial sclerosis.


In view of the widely divergent clinical course that patients with FSGS may follow, an attempt has been made to classify FSGS into distinct histologic subcategories. A scheme that has been proposed includes five variants: Perihilar, tip, cellular, collapsing, and not otherwise specified. Additional studies are necessary to determine whether this categorization provides information that can be used to guide treatment or clarify the long-term prognosis.
Pathogenesis
This form has also been called idiopathic FSGS. Based on clinical evidence of variable response to immunosuppressive medications, the presumption has been that primary FSGS reflects a disturbance in the immune system. However, unlike MCNS, no consistent abnormality has been demonstrated except for altered synthesis and release of tumor necrosis factor (TNF)–α in peripheral blood leukocytes. In addition, various circulating factors including hemopexin, and immunoglobulin-like molecules have been isolated from the sera of patients with FSGS. Removal of these circulating factors, using plasmapheresis or immunoadsorption columns, has been associated with disease remission and infusion into animals has resulted in glomerular proteinuria. Further work on the nature of these substances may help identify the cause of proteinuria in FSGS and define better treatments.
Exciting findings over the past 10 years underscore the pivotal role of the podocyte in maintaining the integrity of the glomerular permselective barrier. A number of proteins have been identified that are components of the cell membrane or actin cytoskeleton of the podocyte. These include nephrin, α-actinin-4, podocin, CD2AP, Wilms tumor suppressor (WT1), and TRPC6 (Figure 25–2). Mutations in the genes for these proteins, occurring in autosomal dominant and recessive patterns, have been associated with steroid-resistant nephrotic syndrome and biopsy-proven FSGS (Table 25–1). Recent series suggest that up to 25% of familial and sporadic cases of steroid-resistant nephrotic syndrome in Europe are related to these genetic abnormalities. Moreover, the response to standard immunosuppressive medications and the risk of recurrent disease after transplantation may be markedly lower in patients with a genetic basis for FSGS. Clarification of these issues is imperative in designing an optimal approach to the evaluation and treatment of patients with FSGS.
Figure 25–2.
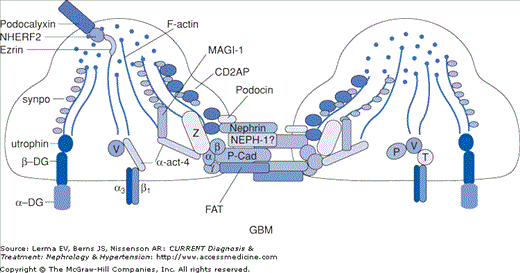
Podocyte architecture. GBM, glomerular basement membrane; synpo, synaptopodin; DG, dystroglycan; α-act-4, α-actinin-4; P-Cad, P-cadherin; P, paxillin; V, vinculin; T, talin. (Reproduced with permission from Mundel P, Shankland SJ: Podocyte biology and response to injury. J Am Soc Nephrol 2002;13:3005.)
Gene Product | Gene | Chromosome | Inheritance |
|---|---|---|---|
Podocin | NPHS2 | 1q25–31 | AR |
α-Actinin-4 | ACTN4 | 19q13 | AD |
CD2AP | CD2AP | 6p12 | AD |
WT-1 | WT-1 | 11p13 | AD or AR |
TRPC6 | TRPC6 | 11q21–22 | AD |
The FSGS lesion represents a nonspecific response to podocyte injury and can arise in a variety of disease states. These include infections with viral agents including HIV and parvovirus B19. A variety of medications including lithium, pamidronate, and illicit drugs such as heroin have been associated with FSGS. Reduced renal mass secondary to surgical ablation (eg, surgery, trauma), reflux nephropathy, and low birth weight can lead to FSGS. Finally, secondary FSGS can occur in patients with a normal renal mass but who have obesity, sickle cell anemia, or cyanotic congenital heart disease (Table 25–2). Identification of these causes and treatment of reversible conditions leads to regression of the FSGS lesions.
Drugs |
Adriamycin |
Heroin |
Interferon-α |
Lithium |
Pamidronate |
Infections |
HIV |
Malarial nephropathy |
Parvovirus B19 |
SV40 virus |
Schistosomiasis |
Malignancies |
Hodgkin’s lymphoma |
Non-Hodgkin’s lymphoma |
Nephron loss |
Reflux nephropathy |
Surgical ablation |
Low birth weight |









