Primary (idiopathic) FSGS
Secondary FSGS
Classic (not otherwise specified) variant
Infection
Tip lesion variant
HIV (usually collapsing variant)
Collapsing glomerulopathy variant
Parvovirus B19
Perihilar variant
Medication and drug related
Familial
Pamidronate (usually collapsing variant)
Alpha-actinin-4 mutation
Interferon alpha
TRPC6 mutation
Lithium
INF2 mutation
Heroin
Podocin mutation
Intravenous drug abuse
Nephrin mutation
Reduced nephron mass (usually perihilar variant)
WT1 mutation
Unilateral renal agenesis
CD2AP mutation
Oligomeganephronia
ApoL1
Reflux interstitial nephritis
Nephrectomy, surgical, traumatic ablation
Solitary kidney, any cause
Hyperfiltration (usually perihilar variant)
Morbid obesity
Sickle cell disease
Cyanotic congenital heart disease
Hypoxic pulmonary disease
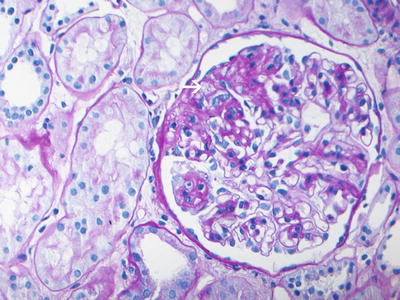
Fig. 4.1
PAS-stained section showing focal and segmental glomerulosclerosis with segmental consolidation of the glomerular tufts, endocapillary foam cells (arrow), and adhesion of the sclerosed segments to the Bowman’s capsule (PAS ×40). Courtesy Sanjeev Sethi, MD
Epidemiology
In adults, membranous nephropathy has traditionally been considered to be the most common histologic lesion underlying the nephrotic syndrome. Prevalences of glomerular disease, however, vary greatly by ethnicity. As a result, reported prevalences of glomerular diseases are difficult to apply to the general population, especially in certain countries with heterogeneous populations such as the USA. Furthermore, studies that report glomerular epidemiological data are inherently limited by changes in renal biopsy practices, changes in disease classification, and referral bias to urban tertiary centers. Nonetheless, there has been an emerging trend over the past three decades showing a disproportionate rise of FSGS in adults compared to other glomerulopathies in Europe, Australia, and the USA, with 15–25 % of nephrotic syndrome explained by this entity [3–5]. The most recent US population-based study using data from the United States Renal Data System (USRDS) and the National Center for Health Statistics showed that FSGS is the most common cause of ESRD due to a primary glomerular disease in both the black and white population during the period of 1980–2000 [6]. The proportion of ESRD due to FSGS increased 11-fold during the 21 year time period. The reason for this trend can be only partially explained by the rising rate of obesity and thus other factors, such as exposure to environmental triggers, need to be considered [7, 8]. Regardless of cause, this is a disturbing trend of clinical importance since FSGS is less amenable to treatment than membranous and carries a poorer renal prognosis.
A rise in the incidence of FSGS has also been observed in children, where minimal change disease is by far the predominant cause of glomerular disease [9]. It is a common clinical practice to empirically treat children with nephrotic syndrome with steroids, an effective treatment for minimal change disease. However, the rise of FSGS in children as well should keep the pediatric clinician mindful of this as a diagnostic possibility, especially in cases of steroid-resistant disease.
Certain studies have recently reported FSGS as the most common cause of primary glomerular disease including reports from Jamaica, Columbia, Brazil, and Jordan [10–13]. Collectively, these epidemiological data highlight the preponderance of FSGS for certain racial groups. Along with this data, observational studies in the USA demonstrate a clear predilection for individuals of African ancestry. The rise of FSGS has been reported to be more marked in African Americans and is 2–3 times more common compared to the European population [6, 14, 15]. Furthermore, nearly all patients with collapsing FSGS due to HIV are black [16]. These observations provide the rationale for population-based genetic studies of African Americans with FSGS, discussed below. Individuals of African ancestry also appear to have more malignant disease, with an earlier age of onset and an increased likelihood of progressing to ESRD. There is a disparity among genders as well, with males constituting the majority of individuals with FSGS [6].
Histologic Variants
FSGS can be a challenging diagnosis due to the nonspecific nature of the histologic findings. Segmental glomerular scarring in some, but not all, of the glomeruli is the signature lesion of FSGS observed on renal pathology, which can be idiopathic or related to a secondary cause (Fig. 4.1, Table 4.1). Furthermore, the pattern of focal and segmental glomerular scarring can also arise from a variety of inflammatory, proliferative, thrombotic, and chronic glomerulonephritides. The Columbia classification, described below, is a morphologic classification for FSGS and is based entirely on the assessment of glomerular light microscopic features, though it also requires immunofluorescence and electron microscopic examination to exclude these other causes of glomerular scarring [17].
Immunofluorescence shows neither immunoglobulin nor complement deposition but focal and segmental deposition of IgM, C3, and C1 can be observed within segmental glomerular sclerosis and hyalinosis. Weak mesangial deposition of IgM and C3 can also be observed. Albumin and immunoglobulins IgA and IgG may be found in the podocytes and in the proximal tubule along with C3. Substantial glomerular deposits of IgG or IgA in mesangial, subendothelial, or subepithelial locations suggest an alternate diagnosis to FSGS.
On electron microscopy, there are changes in the podocyte including hypertrophy, microvillous transformation, detachment from the glomerular basement membrane (GBM), and accumulation of hyaline leading to occlusion of the glomerular capillary lumina. Sparse electron densities may be seen in the mesangium. The presence of sizeable or regular electron-dense deposits in the subepithelial location of nonsclerotic capillaries is also indicative of an alternate diagnosis. The glomerular basement membrane is of normal thickness in the nonsclerotic regions and diffuse thinning should raise the possibility of collagen IV disorders. Most notable, however, is the ultrastructural finding of the so-called foot process fusion, also known as effacement, which is observed on light microscopy as well.
Another diagnostic consideration is the quality of the biopsy sample and specifically the number of glomeruli sampled is particularly important given the focality of lesions. If a lesion that affects only 5 % of glomeruli is to be detected or excluded with 95 % confidence, then over 20 glomeruli are needed in the biopsy [18]. In cases where only a few glomeruli are sampled, minimal change disease may be erroneously diagnosed instead of FSGS.
A standardized classification scheme for FSGS was devised in 2000 at Columbia University, New York, at an international consensus meeting of renal pathologists.
There are multiple examples of consensus classifications as seen in the ISN classification of lupus nephritis, the Chapel Hill classification of systemic vasculitides, and the Banff classification of renal transplant pathology [19–23]. These schemas are formulated with the hopes of reflecting information on stage of disease, prognosis, and response to treatment. However, the significance of the standardized histologic variants in FSGS remains controversial. With that in mind, the Columbia schema defines five main light microscopic patterns of FSGS termed FSGS not otherwise specified (NOS), peripheral variant, cellular variant, tip variant, and collapsing variant (Table 4.2) [17, 18].
Table 4.2
Pathologic criteria for FSGS variants
Histologic variant | Positive criteria | Negative criteria |
|---|---|---|
Collapsing | At least one glomerulus with: (1) Glomerular capillary tuft collapse (2) Overlying podocyte hypertrophy and hyperplasia Features of other variants may be present | |
Tip | At least one glomerulus with: (1) Segmental lesion involving the outer 25 % of the glomerular tuft, otherwise known as the tip domain. Origin of proximal tubule must be identifiable (2) Adhesion or confluence of lesion in the tip domain (3) Foam cells or endocapillary cells in tip lesion or sclerosis | (1) Exclude collapsing variant (2) Exclude perihilar variant |
Cellular | At least one glomerulus with: (1) Endocapillary hypercellularity, typically due to foam cells in any segment of glomerular capillary but involving at least 25 % of the glomeruli (2) Other glomeruli may have segmental sclerosis | (1) Exclude collapsing variant (2) Exclude tip variant |
Perihilar | (1) More than 50 % of glomeruli with segmental lesions must have perihilar sclerosis (2) At least one glomerulus must have perihilar hyalinosis | (1) Exclude collapsing variant (2) Exclude tip variant |
Not other specified | (1) Segmental glomerular capillary tuft lesion (2) May have segmental capillary wall collapse, without podocyte hyperplasia; peripheral or perihilar segmental sclerosis (3) Any number of glomeruli are involved | (1) Exclude all other variants |
FSGS Not Otherwise Specified
FSGS (NOS) is the most common histologic variant. This form is also sometimes referred to as classic FSGS or FSGS of the usual type. The diagnosis of FSGS (NOS) relies on the exclusion of the other variants. On light microscopy, FSGS (NOS) is characterized on by the presence of focal and discrete segmental consolidation of glomerular tufts by sclerosis that obliterates some glomerular capillary lumens. The sclerotic lesions, characterized by increased matrix, affect variable parts of the glomerulus, including both the perihilar and peripheral areas. These changes occur first in the juxtamedullary glomeruli and thus superficial biopsies containing only cortex may miss these lesions. Hyaline deposits may be apparent within the sclerotic lesions, which also may form adhesions to Bowman’s capsule. Podocyte effacement is observed above the sclerotic lesions along with separation from the glomerular basement membrane and intervening accumulation of new matrix leading to “podocyte capping.” The degree of podocyte effacement generally correlates with the severity of proteinuria. Typically, podocyte hypertrophy or hyperplasia is mild in this variant. Mesangial hypercellularity, glomerulomegaly, and arteriolar hyalinosis may occur. Tubular atrophy and interstitial fibrosis are patchy and usually correlate in severity and to the distribution of the sclerosis, though sometimes have been observed to be disproportionately severe, presumably due to conditions of prolonged and heavy proteinuria which mediate tubular injury itself. In some cases, FSGS (NOS) may represent increasing chronicity and be an advanced stage of the other morphologic variants.
FSGS Perihilar Variant
The tip, cellular, and collapsing variants need to be first excluded. To meet criteria for this variant, greater than 50 % of segmentally sclerosed glomeruli must have sclerosis and hyalinosis adjacent to the vascular pole, otherwise known as the perihilar region. Other glomeruli may have randomly distributed lesions identical to those in FSGS (NOS). Glomerulomegaly and adhesions are common findings. Arteriolar hyalinosis is frequently observed and sometimes in contiguity with hyalinosis in the perihilar sclerotic segment. Podocytes may be hypertrophied and hyperplastic, but this is not as pronounced as with the tip, cellular, and collapsing variants. Mesangial hypercellularity is usually minimal or absent.
Though the perihilar variant can be found in primary FSGS, it is an especially common finding in FSGS due to secondary causes, namely, those related to low nephron number and hyperfiltering states. Such causes include obesity, cyanotic congenital heart disease, reflux nephropathy, renal agenesis, dysplasia, oligomeganephronia, or any advanced renal disease with a reduced number of functioning nephrons.
FSGS Cellular Variant
The diagnosis of cellular FSGS requires exclusion of the tip and collapsing morphologic variants. It is defined by the presence of at least one glomerulus with endocapillary hypercellularity involving at least 25 % of the tuft and causing occlusion of the capillary lumina. These cells include endothelial cells, foam cells, and infiltrating leukocytes, such as monocytes, macrophages, and sometimes lymphocytes and neutrophils. Any segment can be affected. Extracapillary hypercellularity is also present, manifesting with podocyte hyperplasia and hypertrophy, which can enlarge and crowd to the degree that they resemble crescents. Adhesions may be present but glomerulomegaly and mesangial hypercellularity are uncommon.
The prognostic significance of this histologic variant is controversial, namely, because the published information regarding clinical course is again limited. Some published literature supports the notion that the cellular variant is characterized by severe proteinuria and represents an early phase in the evolution to segmental sclerosis [24–27]. This is based on the observation that there is a shorter time course from onset of proteinuria to renal biopsy. However, it is also possible these cases are actually collapsing FSGS, given the overlapping characteristic of hypercellularity but misclassified due to limited sampling.
FSGS Tip Variant
To make the diagnosis of the tip variant, the collapsing variant must be excluded. The name describes the location of the segmental lesion in at least one glomerulus—involving the quarter of the tuft adjacent to the origin of the proximal tubule—the glomerular tip. Sometimes, the affected segment may even appear to be herniated into the tubular lumen. Visceral epithelial cells next to the consolidated segment are hypertrophied and swollen, often becoming contiguous and attached to the nearby parietal and tubular epithelial cells at the origin of the proximal tubule. Adhesions between the tuft and Bowman’s capsule occur. These lesions can expand toward the glomerular hilum, though they originate in the periphery.
The tip lesion was first reported in the literature by Howie and Brewer and described to be a steroid sensitive entity, thus resembling minimal change rather than FSGS [28]. Furthermore, autopsies of children in the presteroid era, who died of lipoid nephrosis, a term used at that time to encompass minimal change disease and FSGS, were found to have tip lesions but have no other histologic evidence of FSGS [29]. Based on this data, it is thought that prolonged proteinuria may cause tubular damage, resulting in the tip lesion. However, the data is scant and it is therefore difficult to draw definitive conclusions. A report of patients who had undergone serial biopsies shows an evolution of tip variant to classic FSGS in a subset of those individuals. Patients with tip variant FSGS have also been described to be steroid/calcineurin inhibitor unresponsive and to have recurrence of classic FSGS post transplant [30]. It is possible that the tip variant reflects two conditions, one an early form of classic FSGS and the other closely related to minimal change disease.
Collapsing FSGS
The presence of a collapsing lesion in even just one glomerular tuft is enough to make a diagnosis of collapsing FSGS, preempting all the other diagnostic subcategories. The collapsing lesion can be segmental or global and usually overlies shrunken, collapsed capillaries and a wrinkled glomerular basement membrane. There is a lack of appreciable increase in intracapillary or mesangial matrix, reflecting the acute nature of the injury. Overlying podocytes are markedly hypertrophied and hyperplastic, with enlarged vesicular nuclei, frequent nucleoli, occasional binucleated forms, and rare mitotic figures. Tubular atrophy and interstitial fibrosis can be disproportionately severe to the degree of collapse observed.
Collapsing FSGS is frequently associated with HIV but can occur idiopathically as well. The majority of individuals with HIV-associated collapsing FSGS are black. Endothelial tubular reticular inclusions observed on electron microscopy are not seen in primary disease and suggest concomitant HIV infection. There are other secondary causes of collapsing FSGS including drugs and other infections, which need to be excluded before arriving at a diagnosis of primary FSGS.
Since its original description in the literature in 1986, both primary and secondary collapsing FSGS have been described to have unique clinical and demographic characteristics compared to the other morphologic variants [31]. As with all FSGS, the collapsing variant is seen more in adults and in males. This morphologic variant has a particular preponderance for African Americans but affects the Caucasian population as well. Individuals with the collapsing variant have a more malignant course. Typically, there is a shorter time from onset of proteinuria to biopsy due to the fact that affected individuals present with higher grade proteinuria and more severe features of nephrotic syndrome and are more likely to have renal insufficiency [7, 16]. This morphologic variant also tends to have the most malignant clinical course and is commonly steroid resistant with progression to ESRD.
The Glomerular Filtration Barrier
The kidney filters approximately 180 L/day of plasma containing over 7,200 g of albumin, 99.9 % of which is retained. Plasma is filtered passively, with only the formed elements of the blood and the bulk part of the plasma proteins being retained [32]. Filtration occurs at the glomerular capillary wall, which separates the blood compartment from the urinary space. It consists of three layers; from the innermost layer to the outermost layer, they are the fenestrated endothelium, the glomerular basement membrane, and the podocyte (Fig. 4.2). This filtration barrier behaves as a size-selective sieve restricting the passage of macromolecules on the basis of their size, shape, and charge. Both size and charge influence filtration of macromolecules. Negatively charged molecules are more restricted in their passage due to the negative charge of the glomerular filter and filtration is also inversely proportional to size [33]. Disruption at any layer of the glomerular capillary wall can lead to passage of macromolecules and proteinuria. While it is logical to conceptually divide the glomerular filter into three parts based on structure, it has been difficult to differentiate the physiology of the filter along similar lines. However, as will be discussed below, advances in the biology of proteinuric renal disease suggest that the podocyte and its slit diaphragm are central to barrier function and disruption here can lead to FSGS.
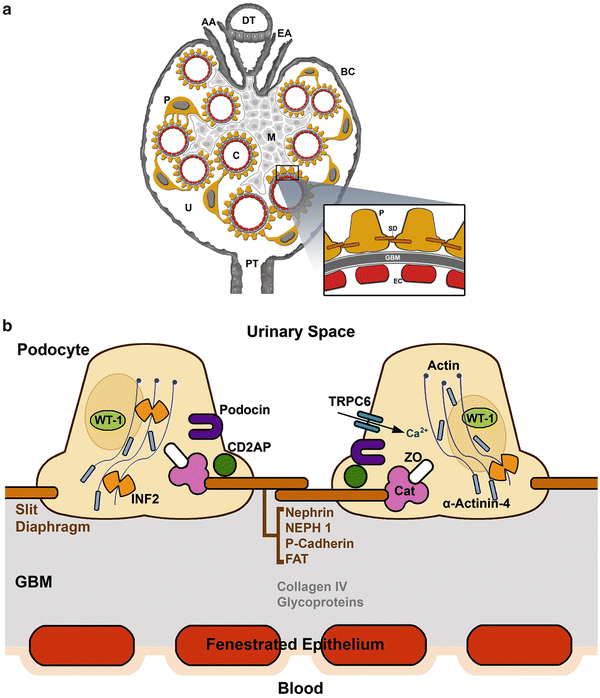
Fig. 4.2
(a) Glomerular filtration barrier structure. (A) Glomerular structure: (AA) afferent arteriole, (EA) efferent arteriole, (DT) distal tubule, (C) capillary loop, (P) podocyte, (FP) podocyte foot processes, (M) mesangium, (U) urinary space, (BC) Bowman’s capsule, and (PT) proximal tubule. Details of the glomerular filtration barrier structure: (FP) podocyte foot process, (GBM) glomerular basement membrane, (EF) endothelial cell, and (SD) slit diaphragm. Inset indicates the glomerular filtration barrier formed by fenestrated endothelial cells, the GBM and podocytes. (b) The podocyte has a unique actin cytoskeleton made up of F-actin and non-muscle myosin such as MYH9 and actin-binding proteins including ACTN4 and INF2. WT1 is a nuclear transcription factor. The slit diaphragm is formed partially by nephrin, podocin, and CD2AP. These molecules at the slit diaphragm associate with TRPC6 and PLCE1
Endothelium
Unlike the endothelial lining of vessels in other parts of the body, the glomerular endothelium is fenestrated with an apparent lack of bridging diaphragms. Therefore, in the past, the endothelium has been viewed as not significantly contributing to protein barrier function. This model is controversial, however, as it theoretically would result in massive convective movement of albumin and other macromolecules into the glomerular basement membrane, causing clogging of the filter [34]. Interestingly, the presence of filamentous plugs filling the capillary fenestrae has been recently described, and it is possible that postmortem artifactual tissue changes explain these apparent discrepancies [35].
In addition, there is now an emerging body of literature to support the concept of cross talk between podocytes, endothelia, and mesangial cells in maintaining glomerular capillary wall function. The production of vascular endothelial growth factor (VEGF) by podocytes is necessary for the integrity of the glomerular endothelium [36]. The upregulation and secretion of the podocyte protein angiopoietin-like 4 (Angptl4) into the glomerular capillary wall cause marked proteinuria in experimental models of nephrotic syndrome [37]. Clinically, VEGF inhibitors used as anti-angiogenic drugs in the treatment of malignancies have been observed to lead to glomerular disease manifesting with proteinuria and thrombotic microangiopathy [36].
The Glomerular Basement Membrane
Of the three layers, the GBM is unique in that it is a specialized extracellular matrix lying between two cellular layers. It is composed of a fibrous network consisting of type IV collagen (collagen α[alpha]3, α[alpha]4, and α[alpha]5 chains), laminin, and nidogen/entactin, together with negatively charged heparin sulfate proteoglycans such as agrin and perlecan. It is the negative charge of the glomerular basement membrane that is traditionally felt to contribute to glomerular permselectivity, though the importance of this in the filtration barrier is debatable [38]. The GBM, however, does account for most of the restriction of fluid flux [39, 40]. Mutations in different components of the GBM lead to varied phenotypes. Human laminin beta2 deficiency gives rise to massive proteinuria but mutations in type IV collagen lead to Alport’s or thin basement membrane disease, in which proteinuria is not a prominent symptom [41].
The Podocyte
Familial FSGS accounts for only a minority of disease, but nevertheless studies of inherited forms of disease have revealed insights into the molecular mechanisms underlying the development of FSGS. Since the discovery of nephrin as the culprit gene for congenital nephrotic syndrome of the Finnish (CNF) type by Karl Tryggvason and colleagues, subsequent studies have consistently pointed to podocyte dysfunction as central to the development of proteinuric kidney disease, including FSGS (Fig. 4.2) [42–49]. More specifically, the cell-cell junction between adjacent podocytes, the slit diaphragm, is made up of a multiprotein complex which dynamically controls foot process architecture via actin cytoskeleton signaling.
The podocyte is a terminally differentiated polarized epithelial cell that stems from precursor mesenchymal cells. During nephrogenesis, these cells undergo modification from a classic epithelial cell phenotype to a highly specialized octopus-shaped cell essential in glomerular filtration. The podocyte consists of a cell body and the major or primary processes, which branch into finger-shaped extensions called pedicles or foot processes. The podocytes cover the external surface of the capillaries, interacting with the GBM and interdigitate through their foot processes in a complex manner forming a network of narrow gaps. The gap spans a space of 30–40 nm and is bridged by a mesh-like network that stretches between these processes called the slit diaphragm. Originally described by Rodewald and Karnovsky in mice, the slit diaphragm forms the glomerular ultrafiltration barrier that allows for the passage of water and solutes while inhibiting loss of larger plasma molecules such as albumin and immunoglobulins [50]. The slit diaphragms (SD) appear early on in nephrogenesis at the capillary stage of development. A modified adherens junction, the SD is a zipper-like structure composed of a complex interplay of a number of proteins including ZO-1, FAT, and nephrin that are all transmembrane proteins shown to localize to the slit diaphragm. ZO-1 localizes to the cytoplasmic face of the SD and interacts with cell junction components as well as proteins in the cytoskeleton [51, 52]. FAT, a member of the cadherin superfamily, localizes at the slit diaphragm [53]. Within the kidney, the integral membrane protein nephrin is expressed exclusively in podocytes [54, 55]. Nephrin-deficient mice and humans develop severe nephrosis [55]. Additional slit diaphragm-associated proteins include podocin and CD2AP which both interact with nephrin and P-cadherin, which is involved in junction-cytoskeleton attachment [56].
The architecture of the podocyte and its unique shape are further characterized by a cytoskeleton composed of microtubules, intermediate filaments, and microfilaments. The cell body cytoskeleton is composed mostly of microtubules and intermediate filaments, with prominent vimentin and desmin. Cytoplasmic extensions or foot process extensions arise from the podocyte cell body. These extensions contain both a microfilament-based contractile apparatus and an actin cytoskeletal network. A group of intracellular cytoskeleton-associated proteins (talin, paxillin, and vinculin) link actin filaments to the cell membrane-associated integrins at the glomerular basement membrane [57].
The role of the podocyte and the slit diaphragm as functional barriers to protein filtration is further defined by its polarity. The exposed surface of the podocyte is covered with a cell coat that contains the sialic acid protein podocalyxin that is thought to give rise to its anionic charge [58]. A decrease in the content of sialic acid has been observed in rats with aminoglycoside nephrosis and in humans with proteinuria or glomerulonephritis [59]. In addition, neutralization of the anionic charge of the sialoprotein coat results in loss of epithelial foot processes that resembles the loss of foot processes seen in human with nephrotic syndrome [60].
FSGS as a Podocyte Disorder
Nearly all the products of genes identified as mutated in nonsyndromic hereditary forms of steroid-resistant nephrotic syndrome (SRNS) and FSGS localize to the podocyte and the slit diaphragm, thereby rationalizing our podocentric view of proteinuric kidney disease (Fig. 4.2c). Furthermore, relative or absolute podocyte number depletion in animal models has been shown to lead to initiation or progression of the lesions observed in FSGS [61–64].
Point mutations in the different genes lead to clinical manifestation of disease at different ages (Table 4.3). Genes that are responsible for recessive forms of nonsyndromic SRNS, which may or may not manifest histologically as FSGS, include nephrin (NPHS1), podocin (NPHS2), laminin-β[beta]2 (LAMB2), and phospholipase C-ε[epsilon]1 (PLCE1). These disorders occur during the first 3 months of life (congenital disease), during infancy (within 1 year), or during childhood. The earlier the onset of SRNS, the more likely that it is of monogenic origin, as 85 % of all congenital SRNS and 66 % of all infantile SRNS is caused by mutations in one of four genes: nephrin, podocin, LAMB2, or WT1 [65]. In the minority of cases that are nongenetic, secondary causes like infections with syphilis and toxoplasmosis or toxin exposure to mercury ought to be ruled out. While those forms of SRNS that present at birth typically do not show evidence of FSGS, it seems appropriate to group these forms of inherited podocyte injury together given their related etiologies.
Table 4.3
Nonsyndromic hereditary causes of FSGS and nephrotic syndrome
Gene | Locus | Inheritance | Protein | Phenotype |
|---|---|---|---|---|
Slit diaphragm | ||||
NPHS1 | 19q13.1 | AR | Nephrin | Congenital nephrotic syndrome of the Finnish type |
NPHS2 | 1q25.2 | AR | Podocin | Congenital-, childhood-, or late-onset SRNS. Juvenile- or adult-onset disease in affected individuals bearing the R229Q mutation along with a pathogenic mutation |
PLCE1 | 10q23.33 | AR | Phospholipase C epsilon 1 | Early-onset SRNS with DMS and FSGS |
CD2AP | 6p12 | AR(?) | CD2 associated protein | Unclear phenotype in humans. Mouse model develops severe proteinuria |
TRPC6 | 11q22.1 | AD | TRPC6 | Adult-onset FSGS |
Actin cytoskeleton | ||||
ACTN4 | 19q13 | AD | Alpha-actinin-4 | Adult-onset FSGS |
INF2 | 14q32.33 | AD | INF2 | Adult-onset FSGS |
Nuclear | ||||
WT1 | 11p13 | AD | Wilms’ tumor 1 | Isolated NS or as part of Frasier or Denys-Drash syndrome |
Glomerular basement | ||||
LAMB2 | 3p21 | AR | Laminin beta-2 | Isolated NS or as part of Pierson syndrome |
Other | ||||
ApoL1 | 22q13.1 | Recessive risk inheritance | Apolipoprotein 1 | Risk haplotypes associated with increased risk of FSGS and ESRD in African Americans |
Point mutations in genes which cause autosomal-dominant nonsyndromic forms of adult-onset FSGS include α[alpha]-actinin-4 (ACTN4), TRPC6, and INF2. Other dominant genes are WT1 and CD2AP which have variable ages for onset of disease, from birth to adulthood. Most recently, two variants in the APOL1 gene have been shown to explain the increased rate of FSGS in individuals of recent African ancestry.
Nephrin (NPHS1)
Mutations in nephrin cause congenital nephrotic syndrome of the Finnish type (CNF), so named because it is most common in Finland but it does exist in other populations. CNF is inherited as an autosomal recessive trait, with both sexes being affected equally and leads to SRNS. Nephrin, encoded by NPHS1, is a transmembrane adhesion protein of the immunoglobulin superfamily, expressed almost exclusively in the podocyte. It forms homodimers and heterodimers with NEPH1 at the slit diaphragm and is involved in podocyte cell signaling to the actin cytoskeleton. To date, close to 200 nephrin mutations have been described in both the Finnish and non-Finnish population. These mutations include small deletions, insertions, nonsense, missense, splice site, and promoter variations and they are distributed throughout the gene [66]. However, 90 % of affected Finnish individuals harbor either the Fin-major (nt121delCT) or Fin-minor (R1109X) mutation due to a founder effect [67–70]. NPHS1 disease generally shows very little phenotypic variation, giving rise to congenital SRNS, but interestingly some NPHS1 mutations have been associated with a milder clinical course characterized by childhood onset of disease [66, 71, 72]. Missense mutations that affect nephrin trafficking to the plasma membrane and functionally act as a null allele result in a more severe phenotype, whereas those mutations in which normal trafficking are preserved results in the milder form of disease.
Most infants with NPHS1 disease are born prematurely with a low birth weight for gestational age. The placenta is markedly enlarged causing flexion deformities of large joints. Nephrotic syndrome is severe with profound urinary protein losses leading to marked edema, susceptibility to bacterial infections, thromboembolic complications, poor nutritional status, and retarded stature as well as hypothyroidism from urinary losses of thyroxine-binding protein. The kidneys are enlarged on ultrasound with loss of the corticomedullary border. No single histologic lesion is pathognomonic for NPHS1 disease. End-stage renal disease typically occurs in childhood, though some affected individuals may have a protracted course.
The treatment is challenging. NPHS1 disease is resistant to immunosuppressive therapy [73]. Treatment is initially supportive and includes albumin infusion, gamma globulin replacement, high protein but low salt diet, vitamin and thyroxine substitution, and close monitoring for infectious and thrombotic complications. Sometimes the complications of nephrotic syndrome are unmanageable and bilateral nephrectomy may be performed to prevent ongoing protein losses to control symptoms. Renal transplantation is curative [73].
Podocin (NPHS2)
Mutations in the NPHS2 gene, encoding for the protein podocin, are a common cause of childhood-onset SRNS. However, screening of cases with congenital NS has shown that a significant proportion is due to mutations in the NPHS2 gene as well [74]. Mutations in podocin that result in nonfunctional and often truncated proteins lead to congenital and infantile disease. Homozygous missense mutations manifest in childhood, whereas individuals who carry one pathogenic NPHS2 mutation along with a common benign non-synonymous polymorphism in the population, the pR229Q variant, typically develop juvenile or adult-onset NS [75, 76]. Podocin is required for proper targeting of nephrin into the slit diaphragm and is expressed exclusively in the podocyte [77].
Wilms’ Tumor (WT1)
Wilms’ tumor suppressor gene (WT1) encodes for the zinc finger transcription factor WT1, which functions both as a tumor suppressor and as a critical regulator of kidney and gonadal development [79]. It is expressed in podocytes and controls cellular functions, such as nephrin expression [80]. WT1 mutations have been associated with three renal-related clinical syndromes, often presenting in the first decade of life [81]: (1) Frasier syndrome, which is characterized by SRNS in childhood with histologic finding of FSGS and often slow progression to ESRD in the second or third decade of life, male pseudohermaphroditism, and a high incidence of gonadoblastomas; (2) Denys-Drash syndrome, which is characterized by infantile SRNS with histologic findings of mesangial sclerosis with rapid progression to ESRD usually by age 4, ambiguous genitalia, and WT; and (3) isolated SRNS. One important aspect of management is the issue of malignancy surveillance and consideration to prophylactic oophorectomies and nephrectomies. The management decisions are particularly challenging in individuals with isolated SRNS. Though quite limited, there is some data to suggest that affected individuals with isolated SRNS and a splice site mutation do not develop malignancy, whereas those with missense or nonsense mutations do [81].
Other Genetic Forms of Congenital Nephrotic Syndrome
Congenital nephrotic syndrome is clinically and genetically heterogeneous and the majority of cases, but not all, can be attributed to mutations in nephrin, podocin, and WT1. Positional cloning in consanguineous families with autosomal recessive inheritance led to the discovery of phospholipase epsilon (PLCE1) as a causative gene for an infantile and childhood form of SRNS called NPHS3 [48]. PLCE1 is expressed in the podocyte. Renal histology varies from diffuse mesangial sclerosis in infants to FSGS in children. Notably and distinct from the other inherited forms of NS and FSGS which are typically resistant to treatment, there have been cases of affected individuals responding to immunosuppressive therapy [81]. Laminin-β[beta]2 is a constituent of the GBM and mutations in its gene were first described to cause congenital NS as part of Pierson syndrome, which also includes distinct ocular anomalies with microcoria. However, there have been reports of isolated NS as well [41, 82].
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


