John W. Foreman
Fanconi Syndrome and Other Proximal Tubule Disorders
The proximal tubule is responsible for the reabsorption of the bulk of a number of solutes, including glucose, amino acids, bicarbonate, and phosphate. This chapter describes a number of disorders, mainly heritable, that affect proximal tubule reabsorption, although Chapters 10 and 12 discuss discuss familial forms of hyperphosphaturia and renal tubular acidosis, respectively.
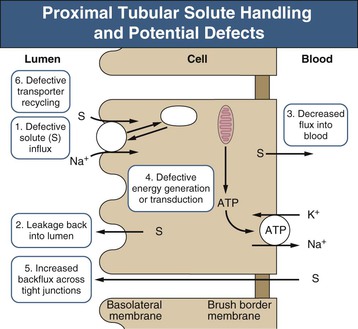
Most nonelectrolyte solutes are reabsorbed in the proximal tubule through specific transport proteins that cotransport them in conjunction with sodium (Fig. 50-1). The driving force for this solute transport is the electrochemical gradient for sodium entry maintained by the enzyme Na+,K+-ATPase. Most disorders of isolated solute reabsorption are related to defects in specific transport proteins, whereas disorders affecting multiple solutes, such as Fanconi syndrome, are probably secondary to defects in energy generation, Na+,K+-ATPase activity, or dysfunction of cellular organelles involved with membrane protein recycling.
Fanconi Syndrome
Definition
In the 1930s, de Toni, Debré, and coworkers and Fanconi independently described several children with the combination of renal rickets, glycosuria, and hypophosphatemia. Fanconi syndrome now refers to a global dysfunction of the proximal tubule leading to excessive urinary excretion of amino acids, glucose, phosphate, bicarbonate, uric acid, and other solutes handled by this nephron segment. These losses lead to the clinical problems of acidosis, dehydration, electrolyte imbalance, rickets, osteomalacia, and growth failure. Numerous inherited or acquired disorders are associated with Fanconi syndrome (Box 50-1).
Etiology and Pathogenesis
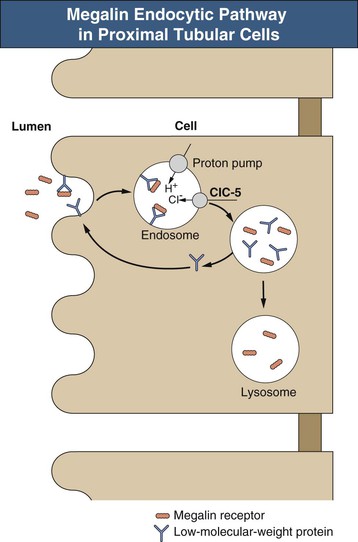
The sequence of events leading to Fanconi syndrome is incompletely defined and probably varies with each cause. Possible mechanisms include widespread abnormality of most or all of the proximal tubule carriers, such as a defect in sodium binding to the carrier or insertion of the carrier into the brush border membrane, “leaky” brush border membrane or tight junctions, inhibited or abnormal Na+,K+-ATPase pump, or impaired mitochondrial energy generation (Fig. 50-1). An abnormality in energy generation has been implicated in a number of disorders, including hereditary fructose intolerance, galactosemia, mitochondrial cytopathies, and heavy metal poisoning, as well as in a number of experimental models of Fanconi syndrome. Abnormal subcellular organelle function, such as the lysosome in cystinosis or the megalin-cubilin endocytic pathway in Dent disease, is also a cause of Fanconi syndrome (Fig. 50-2).
In adults, the most common causes of persistent Fanconi syndrome are an endogenous or exogenous toxin such as a heavy metal, a medication, and a dysproteinemia. In children, the most common persistent cause is an inborn error of metabolism, such as cystinosis. Specific causes of Fanconi syndrome are discussed after a general description of the clinical manifestations and treatment of the syndrome.
Clinical Manifestations
Fanconi syndrome gives rise to a number of clinical abnormalities (Box 50-2).
Aminoaciduria
Aminoaciduria is a cardinal feature of Fanconi syndrome. Virtually every amino acid is found in excess in the urine, thus the term generalized aminoaciduria. There are no clinical consequences, however, because the losses are trivial, 0.5 to 1 g/day, in relation to the dietary intake.
Glycosuria
Glycosuria secondary to proximal tubule dysfunction is another of the cardinal features of Fanconi syndrome and results from impaired tubular reabsorption of glucose. It is often one of the first diagnostic clues. As with aminoaciduria, glycosuria rarely causes symptoms, such as weight loss or hypoglycemia.
Hypophosphatemia
Hypophosphatemia, secondary to impairment in phosphate reabsorption, is a common finding in Fanconi syndrome. Assessment of tubular phosphate handling can be made by measuring the maximum phosphate reabsorption in relation to the glomerular filtration rate (TmP/GFR) on fasting urine and blood samples. Elevated parathyroid hormone (PTH) and low vitamin D levels also may play a role in the phosphaturia of Fanconi syndrome, although these hormonal abnormalities are not always present. A few patients have impaired conversion of 25-hydroxyvitamin D to 1,25-hydroxyvitamin D; metabolic acidosis, another feature of Fanconi syndrome, may also impair this conversion. Another mechanism for the hypophosphatemia is impairment of the megalin-dependent reabsorption and degradation of filtered PTH.1 Unabsorbed PTH then binds to receptors in more distal portions of the proximal tubule, leading to increased endocytosis of apical phosphate transporters and increased phosphaturia. The hypophosphatemia, especially if accompanied by hyperparathyroidism and low 1,25-hydroxyvitamin D levels, often leads to significant bone disease, presenting with pain, fractures, rickets, or growth failure.
Hyperchloremic Metabolic Acidosis
Hyperchloremic metabolic acidosis, another feature of Fanconi syndrome, is a result of impaired bicarbonate reabsorption by the proximal tubule (proximal or type 2 renal tubular acidosis; see Chapter 12). This impaired reabsorption can lead to the loss of more than 30% of the normal filtered load of bicarbonate. As the serum bicarbonate concentration ([HCO3−]) falls, the filtered load falls, and excretion drops such that the serum [HCO3−] usually remains between 12 and 18 mmol/l. On occasion, there is an associated defect in distal acidification, usually in association with longstanding hypokalemia or nephrocalcinosis. Ammoniagenesis is usually normal or increased because of the hypokalemia and acidosis, unless there is an associated impairment in GFR.
Natriuresis and Kaliuresis
Natriuresis and kaliuresis are common in Fanconi syndrome and can give rise to significant, even life-threatening problems. These electrolyte losses are in part related to impaired bicarbonate reabsorption, with the subsequent urinary excretion of sodium and potassium ions with the bicarbonate. In some cases, sodium and potassium losses are so great that metabolic alkalosis and hyperaldosteronism result, simulating Bartter syndrome despite the lowered bicarbonate threshold. The clearance of potassium may be twice that of the GFR, and the resulting severe hypokalemia can cause sudden death.
Polyuria and Polydipsia
Polyuria, polydipsia, and frequent bouts of severe dehydration are common symptoms in young patients with Fanconi syndrome. The polyuria is mainly related to the osmotic diuresis from the excessive urinary solute losses; but some patients have an associated concentrating defect, especially those with prolonged hypokalemia.
Growth Retardation
Growth retardation in children with Fanconi syndrome is multifactorial. Hypophosphatemia, disordered vitamin D metabolism, and acidosis contribute to growth failure, as do chronic hypokalemia and extracellular volume contraction. Glycosuria and aminoaciduria probably do not play a role. However, even with correction of all these metabolic abnormalities, most patients fail to grow, especially those with cystinosis.
Hypouricemia
Hypouricemia, caused by impairment in renal handling of uric acid, is often present in Fanconi syndrome, especially in adults. Urolithiasis from the uricosuria has only rarely been reported, probably because the urine flow and pH are increased, inhibiting uric acid crystallization.
Proteinuria
Proteinuria is usually minimal, except when Fanconi syndrome develops in association with the nephrotic syndrome. Typically, only low-molecular-weight proteins (<30,000 daltons) are excreted, such as vitamin D and A–binding proteins, enzymes, immunoglobulin light chains, and hormones.
Treatment of Fanconi Syndrome
Therapy, whenever possible, should be directed at the underlying causes of Fanconi syndrome (see later discussion). This includes avoidance of the offending nutrient in galactosemia, hereditary fructose intolerance, or tyrosinemia; treatment of Wilson disease with penicillamine and other copper chelators; or treatment of heavy metal intoxication by chelation therapy. In these patients, resolution of Fanconi syndrome usually is complete.
In other patients with Fanconi syndrome, therapy is directed at the biochemical abnormalities secondary to the renal solute losses and at the bone disease often present in these patients. The proximal renal tubular acidosis (type 2 RTA) usually requires large doses of alkali for correction. Some patients benefit from hydrochlorothiazide to minimize the volume expansion associated with these large doses of alkali. Potassium supplementation usually is also needed, especially if there is a significant RTA. The use of potassium citrate, lactate, or acetate will correct not only the hypokalemia but also the acidosis. A few patients will require sodium supplementation along with potassium. Again, the use of a metabolizable anion will aid in the correction of the acidosis. Rarely, patients may need sodium chloride supplementation. Usually, these patients have alkalosis when untreated as a result of large urinary NaCl losses, which lead to volume contraction that overrides the RTA. Magnesium supplementation may be required. Adequate fluid intake is essential. Correction of hypokalemia and its effect on the concentrating ability of the distal tubule may lessen the polyuria.
Bone disease is multifactorial, including hypophosphatemia, urinary loss of vitamin D–binding protein and vitamin D, decreased synthesis of calcitriol in some patients, hypercalciuria, and chronic acidosis. Hypophosphatemia should be treated with 1 to 3 g/day of oral phosphate with the goal of normalizing serum phosphate levels. Many patients with Fanconi syndrome will require supplemental vitamin D for adequate treatment of the rickets and osteomalacia. It is unclear whether standard vitamin D (calciferol [ergocalciferol]) or a vitamin D metabolite is better for supplementation. Currently, most clinicians use a vitamin D metabolite, such as 1,25-dihydroxycholecalciferol (calcitriol). These metabolites obviate the concern of inadequate vitamin D hydroxylation by the proximal tubule mitochondria and reduce the risk for prolonged hypercalcemia because of their shorter half-life. Vitamin D therapy will also improve the hypophosphatemia and lessen the risk for hyperparathyroidism. Supplemental calcium is indicated in those with hypocalcemia after supplemental vitamin D is started. Hyperaminoaciduria, glycosuria, proteinuria, and hyperuricosuria usually do not lead to clinical difficulties and do not require specific treatment. Carnitine supplementation, to compensate for the urinary losses, may improve muscle function and lipid profiles, but the evidence is inconsistent.
Inherited Causes of Fanconi Syndrome
Cystinosis
Definition
Cystinosis, or cystine storage disease, is characterized biochemically by excessive intracellular storage, particularly in lysosomes, of the amino acid cystine.2 Three different types of cystinosis can be distinguished based on clinical course and age at onset and the intracellular cystine content. Benign or adult cystinosis is associated with cystine crystals in the cornea and bone marrow only, as well as the mildest elevation in intracellular cystine levels; no renal disease occurs. Infantile or nephropathic cystinosis is the most common form of cystinosis and is associated with the highest intracellular levels of cystine and the earliest onset of renal disease. In the intermediate or adolescent form, intracellular cystine levels are between those of the infantile and adult forms, with later onset of renal disease.
Etiology and Pathogenesis
Nephropathic cystinosis is transmitted as an autosomal recessive trait localized to the short arm of chromosome 17, with an estimated incidence of 1 in 200,000 live births. The CTNS gene codes for a lysosomal membrane protein, cystinosin, that mediates the transport of cystine from the lysosome.3 The benign and intermediate forms of cystinosis are also associated with CTNS mutations but still have some functional transport protein, leading to lower intracellular cystine levels and slower onset of renal disease in the intermediate form and no renal disease in the benign form.
Clinical Manifestations
The first clinical symptoms and signs in nephropathic cystinosis are those of Fanconi syndrome and usually appear in the second half of the first year of life. Subtle abnormalities of tubular function can be demonstrated earlier in families with index cases, but there always is a delay between birth and the first symptoms. Rickets is common after the first year of life, along with growth failure. The growth failure occurs before the GFR declines and despite correction of electrolyte and mineral deficiencies. The GFR invariably declines and end-stage renal disease (ESRD) occurs by late childhood.
Nephrocalcinosis is relatively common, and a few patients have developed renal calculi. Photophobia is another common symptom that occurs by 3 years of age and is progressive. Older patients with cystinosis may develop visual impairment and blindness. Children with cystinosis usually have fair complexions and blond hair, but dark hair has been observed in some. Cystinosis has been observed in other ethnic groups but is less common than in Caucasians.
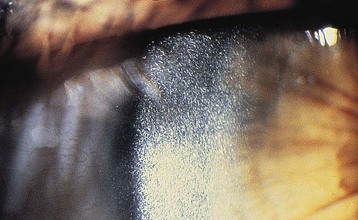
The diagnosis is based on the demonstration of elevated intracellular levels of cystine, usually in white blood cells or skin fibroblasts. Patients with nephropathic and intermediate cystinosis have intracellular cystine levels that exceed 2 nmol half-cystine per mg protein (normal <0.2 nmol half-cystine per mg protein). Heterozygotes for cystinosis will have levels that range from 0.2 to 1 nmol half-cystine/mg protein. A slit-lamp demonstration of corneal crystals strongly suggests the diagnosis2 (Fig. 50-3). A prenatal diagnosis can be made with amniocytes or chorionic villi.
Common late complications of cystinosis include hypothyroidism, splenomegaly and hepatomegaly, decreased visual acuity, swallowing difficulties, pulmonary insufficiency, and corneal ulcerations.5 Less frequently, older patients have developed insulin-dependent diabetes mellitus, myopathy, and progressive neurologic disorders. Decreased brain cortex has also been noted on imaging in some patients. Older patients may develop vascular calcification, especially of the coronary arteries, which can lead to myocardial ischemia.
Pathology
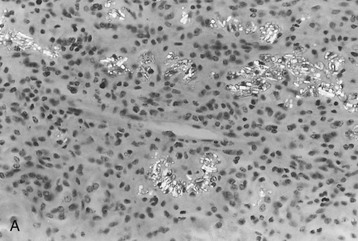
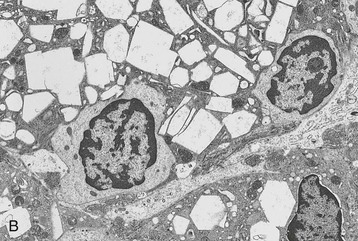
The morphologic features of the kidney in cystinosis vary with the stage. Early in the disease, cystine crystals are present in tubular epithelial cells, interstitial cells, and rarely glomerular epithelial cells6,7 (Fig. 50-4, A). A swan-neck deformity or thinning of the first part of the proximal tubule is an early finding but is not unique to cystinosis. Later, there is pronounced tubular atrophy, interstitial fibrosis, and abundant crystal deposition with giant cell formation of the glomerular visceral epithelium, segmental sclerosis, and eventual glomerular obsolescence. Electron microscopy (EM) studies have demonstrated intracellular crystalline inclusions consistent with cystine (Fig. 50-4, B). Peculiar “dark cells,” unique to the cystinotic kidney, have also been observed.
Treatment
Nonspecific therapy for infantile cystinosis consists of vitamin D therapy and replacement of the urinary electrolyte losses, followed, in due course, by the management of the progressive renal failure (Table 50-1). Cysteamine therapy has now been shown to lower tissue cystine levels and slow the decline in GFR, especially in children with a normal serum creatinine concentration treated before 2 years of age8 (Fig. 50-5). Cysteamine therapy also improves linear growth but not Fanconi syndrome. The most common problems associated with cysteamine therapy are nausea, vomiting, and a foul odor and taste. Treatment should begin with a low dose of cysteamine soon after the diagnosis is made, increased during 4 to 6 weeks to 60 to 90 mg/kg/day in four divided doses as close to every 6 hours as possible. Slowly increasing the dose minimizes the risk for neutropenia, rash, and arthritis. Leukocyte cystine levels should be checked every 3 to 4 months to monitor effectiveness and compliance, with the goal of achieving and maintaining a cystine level of below 2.0 and preferably below 1.0 nmol half-cystine/mg protein. A long-acting formulation of cysteamine should be available soon that will allow twice-daily dosing. A 50-mM solution of cysteamine applied topically onto the eye has proved useful in depleting the cornea of cystine crystals, but it requires administration 6 to 12 times a day to be effective.
Table 50-1
Treatment of cystinosis.
| Treatment of Cystinosis | |
| Problem | Therapy |
| Removal of lysosomal cystine | Cysteamine, 0.235 g/m2 q6h to maintain leukocyte cystine level <1 nmol half-cystine*/mg protein |
| Correction of Tubulopathy | |









