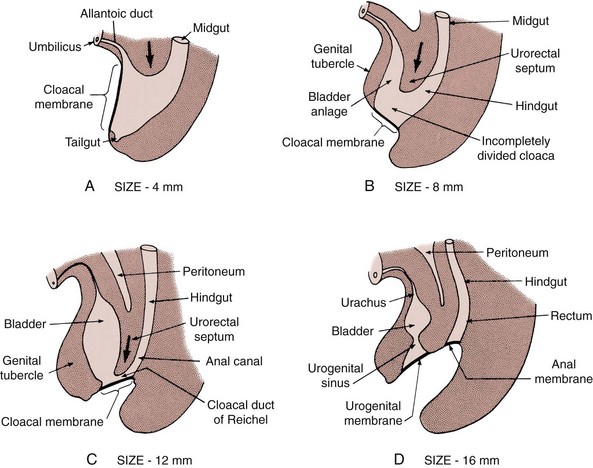
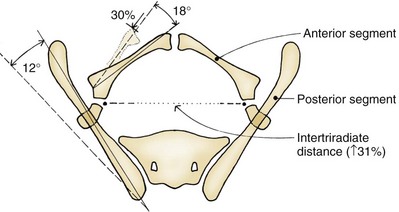
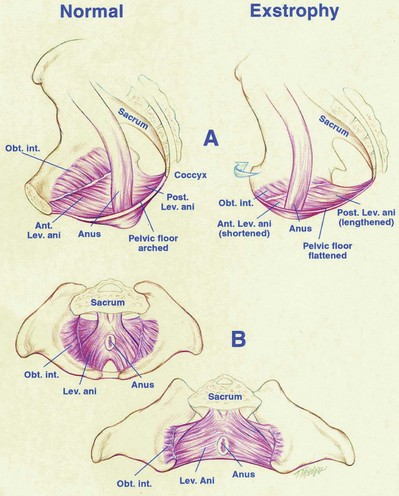
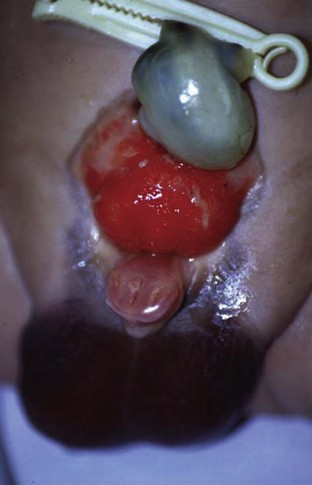
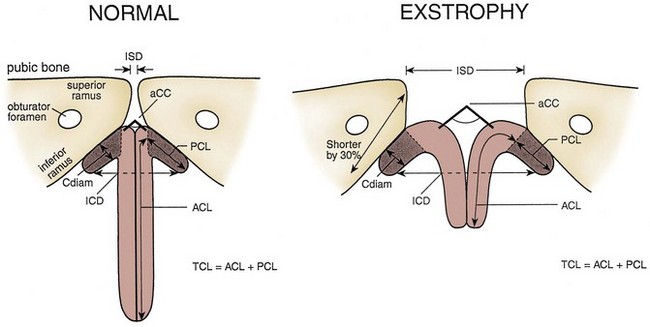
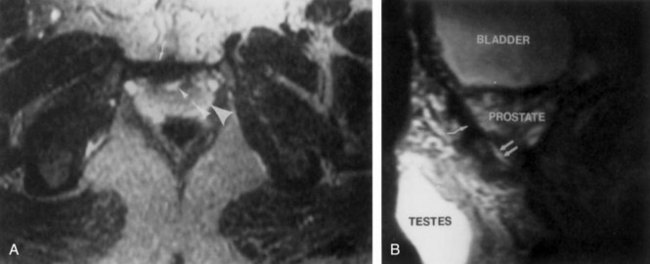
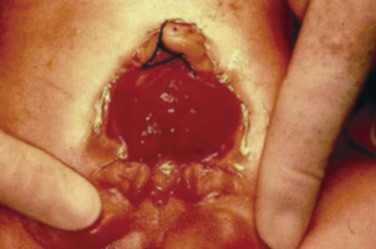
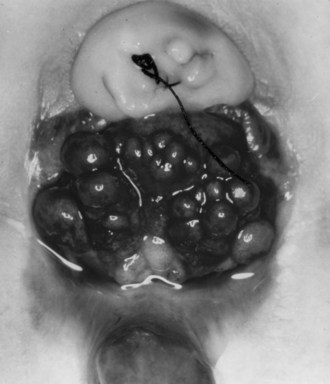
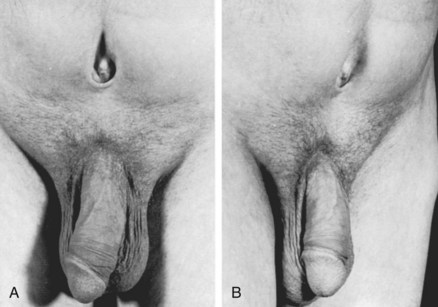
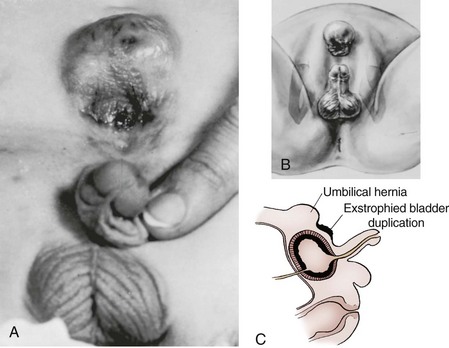
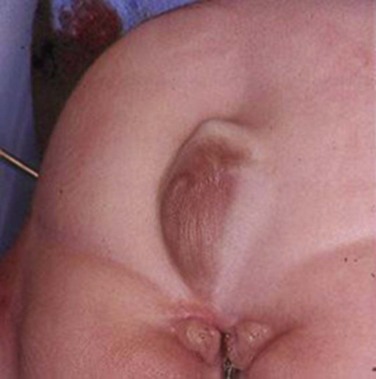
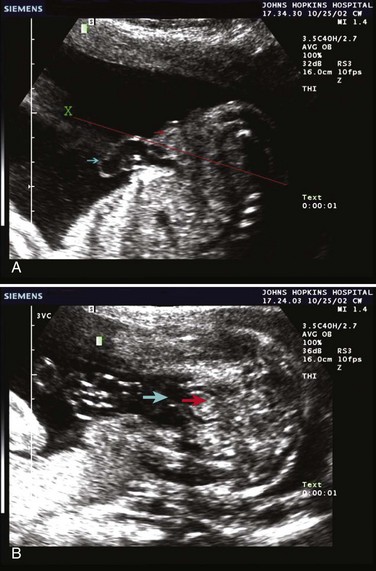
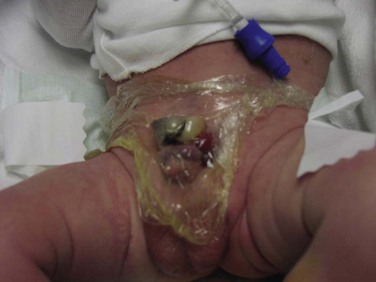
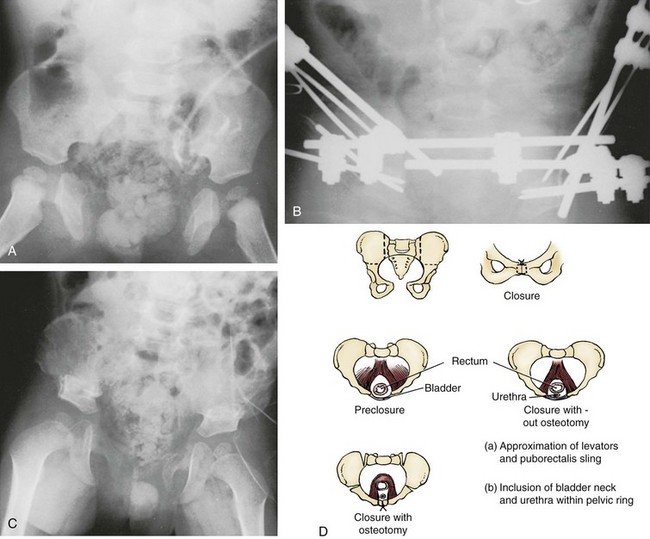
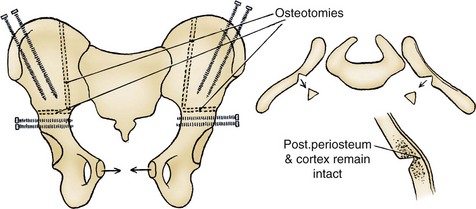
John P. Gearhart, MD, Ranjiv I. Mathews, MD The exstrophy-epispadias complex of genitourinary malformations can be as simple as a glanular epispadias or as overwhelming as a multisystem defect such as cloacal exstrophy (Fig. 124–1). This chapter provides a comprehensive overview of the entire spectrum of the exstrophy-epispadias-cloacal exstrophy defects. In addition, all modern methods of exstrophy management, their complications, and outcomes are discussed. In older texts, the first account of bladder exstrophy was ascribed to Assyro-Babylonian sources dating from the first and second millennia BC. At that time, birth anomalies in both humans and animals were carefully recorded on tablets for their importance as omens on the basis of their interpretation by divination experts. Feneley and Gearhart (2000) examined Assyro-Babylonian descriptions of congenital anomalies from cuneiform texts at the British Museum in London. Although references to anomalies involving the external genitalia were frequent (e.g., hermaphroditism, absence of external genitalia, unilateral and bilateral undescended testes), references to renal and bladder anomalies were few and difficult to interpret medically. Duplication and laterality of anomalies were described in detail owing to their distinct significance, but malformations in combination were not recorded. On the basis of these studies performed with a prominent Assyriologist, a definitive description of bladder or cloacal exstrophy was not corroborated. The first recorded case of epispadias is attributed to the Byzantine Emperor Heraclius (AD 610-641) and the first description of bladder exstrophy to Schenck in 1595 (Feneley and Gearhart, 2000). The incidence of bladder exstrophy has been estimated as between 1 in 10,000 and 1 in 50,000 (Lattimer and Smith, 1966) live births. However, data from the International Clearinghouse for Birth Defects monitoring system estimated the incidence to be 3.3 cases in 100,000 live births (Lancaster, 1987). The male-to-female ratio of bladder exstrophy derived from multiple series is 2.3 : 1 (Shapiro et al, 1985). However, two series reported a 5 : 1 to 6 : 1 male-to-female ratio of exstrophy births (Ives et al, 1980; Lancaster, 1987). The risk of recurrence of bladder exstrophy in a given family is approximately 1 in 100 (Ives et al, 1980). Shapiro and colleagues (1985) conducted a questionnaire of pediatric urologists and surgeons in North America and Europe and identified the recurrence of exstrophy and epispadias in only 9 of approximately 2500 indexed cases. Lattimer and Smith (1966) cited a set of identical twins with bladder exstrophy and another set of twins in whom only one child had exstrophy. Shapiro’s series identified five sets of male and female nonidentical twins in whom only one twin was affected with exstrophy; five sets of male identical twins in whom both twins were affected; one set of identical male twins in whom only one twin was affected; and three sets of female identical twins in whom only one twin had the exstrophy anomaly (Shapiro et al, 1985). Evidence by Reutter and colleagues has demonstrated six families with two occurrences of exstrophy-epispadias complex, one in which the proband was the product of a consanguineous union and four discordant twin pairs. Also, Boyadjiev and colleagues (2004a) found four multiplex families (2.7%) in a cohort of 151 families with the exstrophy-epispadias complex. There were three twin pairs, two of which were monozygotic, and concordance was present in only one of the twin pairs. Consanguinity was present in one family. Bladder exstrophy or epispadias was not reported until the 1980s in the offspring of parents with the exstrophy-epispadias complex. Shapiro and colleagues (1985) described two women with complete epispadias who gave birth to sons with bladder exstrophy. They also reported that another woman with bladder exstrophy gave birth to a son with bladder exstrophy. The inheritance of these three cases of bladder exstrophy was identified in a total of 225 offspring (75 boys and 150 girls) produced by individuals with bladder exstrophy and epispadias. Shapiro and colleagues (1985) determined that the risk of bladder exstrophy in the offspring of individuals with bladder exstrophy and epispadias is 1 in 70 live births, a 500-fold greater incidence than in the general population. Boyadjiev and colleagues (2004a) studied sibling data from 200 families and found 259 unaffected children in addition to the probands with exstrophy. Twenty-six probands had first-, second-, or third-degree relatives with congenital anomalies unrelated to the exstrophy-epispadias complex, most of which were midline defects and oral clefts. Four probands had a total of seven biologic children that were unaffected. In a multinational review of exstrophy patients (Lancaster, 1987), two interesting trends were found: (1) Bladder exstrophy tends to occur in infants of younger mothers, and (2) an increased risk at higher parity is observed for bladder exstrophy but not for epispadias. Boyadjiev’s (2004a) data, however, differ from these trends, indicating that the average maternal age was 34 years and the average paternal age was 32 years. In addition, 49% of probands were born from first pregnancies. This change may represent societal changes, indicating advancing maternal age for first pregnancies and increasing utilization of assisted reproductive techniques in first pregnancies. Exploration of possible etiologies for the exstrophy-epispadias complex continues. A report from Israel indicated a 10-fold increase in exstrophy births to mothers who had received large doses of progesterone in the early part of the first trimester. Wood and colleagues (2003) reported on a sizable series of children with exstrophy conceived using assisted reproductive techniques. A 7.5-fold increase in incidence was noted when in vitro fertilization was used. These two reports indicate a role for hormonal changes in the etiology of the exstrophy-epispadias complex. In a large series of 214 females, no association of parental age, maternal reproductive history, or periconceptual material exposure to alcohol, drugs, radiation, or infections was found (Gambhir et al, 2008). However, periconceptual maternal exposure to smoking was significantly more common in patients with cloacal exstrophy than in the combined group of patients with classic exstrophy/epispadias. Periconceptual use of folic acid offered no protection to this anomaly (Gambhir et al, 2008). Genetic studies to identify a genetic locus for the exstrophy-epispadias complex are under way. Boyadjiev and colleagues (2004b) have reported finding a breakpoint disruption in the 5′ region of the CASPR3 gene on chromosome 9. This observation is the first to suggest a possible genetic basis for development of the exstrophy-epispadias complex. In another recent study by Ludwig and colleagues (2009), risk loci for the exstrophy spectrum were found in seven separate chromosomes with susceptibility genes in several regions. Bladder exstrophy, cloacal exstrophy, and epispadias are variants of the exstrophy-epispadias complex (see Fig. 124–1). The cause of this complex is thought to be the failure of the cloacal membrane to be reinforced by ingrowth of mesoderm (Muecke, 1964). The cloacal membrane is a bilaminar layer situated at the caudal end of the germinal disk that occupies the infraumbilical abdominal wall. Mesenchymal ingrowth between the ectodermal and endodermal layers of the cloacal membrane results in formation of the lower abdominal muscles and the pelvic bones. The cloacal membrane is subject to premature rupture, and, depending on the extent of the infraumbilical defect and the stage of development during which the rupture occurs, bladder exstrophy, cloacal exstrophy, or epispadias results (Ambrose and O’Brien, 1974). After mesenchymal ingrowth occurs, the urorectal septum grows in a caudal direction and divides the cloaca into a bladder anteriorly and a rectum posteriorly (Fig. 124–2). Distally, the septum meets the posterior remnant of the bilaminar membrane, which eventually perforates and forms the urogenital and anal openings. The paired genital tubercles migrate medially and fuse in the midline, cephalad to the dorsal membrane before perforation. The theory of embryonic maldevelopment in exstrophy held by Marshall and Muecke (1968) is that the basic defect is an abnormal overdevelopment of the cloacal membrane, which prevents medial migration of the mesenchymal tissue and proper lower abdominal wall development. The timing of the rupture of this defective cloacal membrane determines the variant of the exstrophy-epispadias complex that results. This theory is supported by Muecke’s work in the chick embryo and by the high incidence of expected central perforations, resulting in the preponderance of classic exstrophy variants. Classic exstrophy accounts for more than 50% of the patients born with this complex (Muecke, 1964; Marshall and Muecke, 1968). Martinez-Frias and colleagues (2001) using epidemiologic factors of low birth weight, twinning, single umbilical artery, and associated defects postulated that cloacal exstrophy and exstrophy of the bladder are two different expressions of a primary developmental field defect with cloacal exstrophy being an early defect. New theories have been offered regarding the origins of these ventral body wall defects. It has been postulated that failure of one or both of the lateral body wall folds to move far enough ventrally to meet its counterpart in the midline (Sadler and Feldkamp, 2008). The associate defect results in one or more of the internal organs protruding through the defective region. Thus if closure fails in the abdominal and pelvic region, cloacal exstrophy results. If in the pelvis alone, only classic exstrophy occurs. Other plausible theories concerning the cause of the exstrophy-epispadias complex exist. Abnormal development of the genital hillocks caudal to the normal position, with fusion in the midline below rather than above the cloacal membrane, has been embraced by other investigators (Patton and Barry, 1952; Ambrose and O’Brien, 1974). Another interesting hypothesis that remains controversial describes an abnormal caudal insertion of the body stalk, which results in a failure of interposition of the mesenchymal tissue in the midline (Mildenberger et al, 1988). As a consequence of this failure, translocation of the cloaca into the depths of the abdominal cavity does not occur. A cloacal membrane that remains in a superficial infraumbilical position represents an unstable embryonic state with a strong tendency to disintegrate (Johnston and Kogan, 1974). The lack of ability of the Meucke hypothesis to explain the absence of the ileocecal region of the digestive system and the superior mesenteric blood supply of the exstrophied intestinal region has led to a suggestion that there may be involvement of the allantois in the development of cloacal exstrophy (Zarabi and Rupani, 1985). Absence of migration, ascent, or alignment of the allantois with the yolk sac with its persistence at the dome of the cloaca can be used to explain the bowel abnormalities noted in cloacal exstrophy. Maldevelopment of the bony pelvis rather than soft tissue defects has been suggested to be the inciting issue for the development of exstrophy. Beaudoin and colleagues (1997) have suggested that lack of “rotation” of the pelvic ring primordium prevents structures attached to the pelvic ring from joining in the midline, allowing herniation of the bladder to occur. The cause of this inadequate rotation remains elusive. Stec and colleagues (2003) were able to demonstrate that the anatomic microstructure of the pelvic ring in exstrophy was similar to that in age-matched control subjects. Formerly, classic bladder exstrophy was thought only to show the characteristic widening of the pubic symphysis caused by malrotation of the innominate bones in relation to the sagittal plane of the body along both sacroiliac joints. In addition, there was an outward rotation or eversion of the pubic rami at their junction with the iliac bones. More recently, Sponseller and colleagues (1995), using computed tomography (CT) of the pelvis with three-dimensional (3D) reconstruction, have further characterized the bone defect associated with both classic bladder exstrophy and cloacal exstrophy. In reviewing a large group of patients with exstrophy of the bladder, using pelvic CT scans and age-matched controls, Sponseller and colleagues (1995) found that patients with classic bladder exstrophy have a mean external rotation of the posterior aspect of the pelvis of 12 degrees on each side, retroversion of the acetabulum, and a mean 18 degrees of external rotation of the anterior pelvis, along with 30% shortening of the pubic rami, in addition to the previously described diastasis of the symphysis pubis (Fig. 124–3). In long-term follow-up there was a foot progression angle of 20 to 30 degrees of external rotation beyond the normal limits seen in early childhood, but this rotation improved with age. Likewise, patients with cloacal exstrophy not only had pelvic deformities to a greater degree but also had asymmetry of the preceding parameters between the right and left sides of the pelvis, malformation of the sacroiliac joints, and occasional dislocations of the hip (Sponseller et al, 1995). Data from Stec and colleagues (2001a) using 3D models from CT scans have provided further insight into the bone defect of children born with classic bladder exstrophy. Using age-matched controls, they found that the sacroiliac joint angle (before closure) was 10 degrees larger in the exstrophy pelvis, being 10 degrees more toward the coronal plane than the sagittal (Fig. 124–4). Also, the bony pelvis in exstrophy has 14.7 degrees more inferior rotation than in healthy patients. Lastly, the sacrum in exstrophy patients has a 42.6% larger volume and 23.5% more surface area than in controls. These new findings will help with planning better osteotomies and better reduction of both the pubic diastasis and pubic long-term undergrowth in these patients. (From Stec AA, Pannu HK, Tadros YE, et al. Evaluation of the bony pelvis in classic bladder exstrophy using 3D CT—further insights. Urology 2001;58:1030.) These rotational deformities of the pelvic skeletal structures contribute to the short, pendular penis seen in bladder exstrophy. Outward rotation and lateral displacement of the innominate bones also account for the increased distance among the hips, waddling gait, and outward rotation of the lower limbs in these children, which in itself causes little disability and usually corrects to some degree over time. Data from Sponseller and colleagues (2001) show an increased incidence of premature osteoarthritis of the hip in adult patients who were closed without an osteotomy. Although these findings do not affect function, periodic radiologic evaluation of the pelvis and lumbosacrum should be performed in adult life. The 30% shortage of bone in exstrophy pelvis has largely gone unexplained. Data by Stec and colleagues (2003) using sections from the bony pelvis in fetal exstrophy and normal aborted fetuses found that the ultrastructure, bone development, microscopic growth patterns, and endochondrial ossification were absolutely the same. Thus restoration of the physiologic shape of the pelvis would likely lead to more normal gross bone growth, decreases in shortage of bone, and a more appropriate distribution of the mechanical and development forces on a more closed normally functioning pelvic ring. Spinal abnormalities have frequently been noted in children with cloacal exstrophy, but incidence in bladder exstrophy has not been studied. A study of 299 children with bladder exstrophy indicated spinal variations without clinical significance (spina bifida occulta, lumbarization or sacralization of vertebrae) in 11%, uncomplicated scoliosis in 2.7%, and spinal dysraphism in 4% including myelomeningocele, lipomeningocele, scimitar sacrum, and hemivertebrae, but only one patient demonstrated any evidence of neurologic dysfunction (Cadeddu et al, 1997). Thus although present in classic bladder exstrophy, symptomatic spinal anomalies appear rare. Stec and colleagues (2001b), using 3D models created from CT scans of children with classic bladder exstrophy and normal age-matched controls, found that the puborectal slings were supporting two times more body cavity area than normal. The levator ani group is positioned more posteriorly in exstrophy patients, with 68% located posterior to the rectum and 32% anterior (vs. 52% posterior and 48% anterior in healthy controls) (Fig. 124–5). The levators are also rotated outward 15.5 degrees, and in the coronal aspect the levators are 31.7 degrees more flattened than normal. This deviation from normal makes the exstrophy puborectal sling more flattened than its normal conical shape. There was no significant difference in the length or thickness of these muscles between patients with exstrophy and normal controls. These new insights further elucidate the entire exstrophy defect, especially in regard to pelvic reconstruction. Interest in development of continence after primary closure in some patients has led to further investigation of the pelvic floor anatomy. Differences in orientation and anatomy of the pelvic floor musculature may directly affect development of continence following closure alone. The successful application of 3D magnetic resonance imaging (MRI) in anorectal anomalies has led to its use for the anatomic definition of the pelvic floor musculature in exstrophy. A comparison of 3D MRI in children with exstrophy before closure and in normal controls indicated that the levator ani group was less dome shaped and more irregular in those with exstrophy (Williams et al, 2004). Also, there was no relationship between the amount of pubic diastasis and the extent of disproportionate curvature of the levator ani group. In addition, Halachmi and colleagues (2003) reported on the postoperative appearance of the pelvic floor with 3D MRI. In two patients who had some degree of continence, the intrasymphyseal distance was noted to be shortest, the angle of the levator ani divergence sharpest, and the bladder neck most deeply positioned in the pelvis. Gargollo and colleagues (2005), reporting on a series of patients who had MRI before and after exstrophy closure, noted that the puborectalis angle in those with dry intervals was decreased compared with that before closure. These two studies correlate well with earlier findings of Gearhart and colleagues (1993c) showing that in the adult patients who were dry, the puborectalis angle was less than 65 degrees. These data reinforce the necessity for aggressive dissection and posterior placement of the posterior urethra and bladder and the role for osteotomy and pelvic fixation. The triangular defect caused by the premature rupture of the abnormal cloacal membrane is occupied by the exstrophy bladder and posterior urethra. The fascial defect is limited inferiorly by the intrasymphyseal band, which represents the divergent urogenital diaphragm. This band connects the bladder neck and posterior urethra to the pubic ramus on anatomic study. The anterior sheath of the rectus muscle has a fanlike extension behind the urethra and bladder neck that inserts into the intrasymphyseal band. Investigations into the relationship of the rectus muscle and fascia to the urogenital diaphragm (Wakim and Barbet, 2002) have found no gross or histologic evidence of the presence of the striated sphincter. However, clear evidence of bladder musculature extending laterally to the pubis was found where it interdigitates with fibers from the rectus fascia forming the fibrous urogenital diaphragm (Wakim and Barbet, 2002). Gearhart and colleagues (1991) have shown the importance of radical incision of these fibers lateral to the urethral plate down to the level of the inferior pubic ramus and levator hiatus for the bladder and posterior urethra to be placed in a deep pelvic position. Data from failed exstrophy closures show these fibers to be intact in many patients at the time of reclosure. The frequent occurrence of indirect inguinal hernias is attributed to a persistent processus vaginalis, large internal and external inguinal rings, and lack of obliquity of the inguinal canal. Connolly and colleagues (1995), in a review of 181 children with bladder exstrophy, reported inguinal hernias in 81.8% of boys and 10.5% of girls. At the time of closure of the bladder exstrophy, these hernias should be repaired by excision of the hernial sac and repair of the transversalis fascia and muscle defect to prevent recurrence or a direct inguinal hernia. The contralateral side should also be explored because the incidence of synchronous or asynchronous bilaterality is high. The divergent levator ani and puborectalis muscles and the distorted anatomy of the external sphincter contribute to varying degrees of anal incontinence and rectal prolapse. Anal continence is usually imperfect at an early age. In rare patients, the rectal sphincter mechanism may never be adequate to control the liquid content of the bowel. Rectal prolapse frequently occurs in untreated exstrophy patients with a widely separated symphysis. It is usually transient and easily reduced. Prolapse virtually disappears after bladder closure or cystectomy and urinary diversion. The appearance of prolapse in an infant is an indication to proceed with definitive management of the exstrophied bladder. If rectal prolapse occurs at any time after exstrophy closure, posterior urethral/bladder outlet obstruction should be suspected and immediate evaluation of the outlet tract by cystoscopy should be performed (Baker and Gearhart, 1998). The male genital defect is severe and is probably the most troublesome aspect of the surgical reconstruction, independent of the decision whether to treat with modern staged closure, combined closure, or a form of urinary diversion (Fig. 124–6). Formerly, it was thought that the individual corpora cavernosa were of normal caliber but appeared shorter because of the wide separation of the crural attachments, the prominent dorsal chordee, and the shortened urethral groove. However, Silver and colleagues (1997b) described the genital defect for the first time in bladder exstrophy in greater detail. MRI was used in adult men with bladder exstrophy and compared with results for age- and race-matched controls. They found that the anterior corporal length of male patients with bladder exstrophy was almost 50% shorter than that of normal controls (Fig. 124–7). However, although the posterior length of the corporal body was the same as in age-matched controls, the diameter of the posterior corporal segment was greater than in normal controls. It was also found on MRI that the diastasis of the symphysis pubis increased the intrasymphyseal and intercorporal distances, but the angle between the corpora cavernosa was unchanged because the corporal bodies were separated in a parallel fashion. Therefore the penis appears short not only because of the diastasis of the pubic symphysis, as thought in the past, but also because of marked congenital deficiency of anterior corporal tissue (Silver et al, 1997b). In a recent surgical anatomic study by Perovic and Djinovic (2007), precise description of the penile defect included (1) corporal bodies separated and triangular in shape; (2) a long convex ventral surface and a short wedge-shaped dorsal surface; and (3) neurovascular bundle length determined by its lie on the individual corporal bodies. In a study by Gearhart and colleagues (1993c), 13 adult men born with bladder exstrophy were evaluated with MRI of the pelvis to evaluate the size and configuration of the prostate and sex accessory organs. The volume, weight, and maximum cross-sectional area of the prostate appeared normal compared with published control values (Fig. 124–8). However, in none of the patients did the prostate extend circumferentially around the urethra, and the urethra was anterior to the prostate in all patients. Except for studies to document the presence of the prostate gland or its size, data do not exist concerning the function of the prostate gland in the patient with exstrophy. Silver and colleagues (1997a) reported free and total prostate-specific antigen levels for a group of adult men with bladder exstrophy. Although they were measurable, they were below the upper limits of established age-specific reference ranges for normal men. The vas deferens and ejaculatory ducts are normal in the exstrophy patient, provided they are not injured iatrogenically. Also, the mean seminal vesicle length in men with exstrophy was normal compared with published controls. Additional evaluation of the puborectalis muscle group was undertaken, and these muscles were found to be widely separated and provided lateral support of the prostate with a narrower puborectalis angle in those who were continent and had undergone prior iliac osteotomy. Autonomic innervation of the corpus cavernosum is provided by the cavernous nerves. These autonomic nerves are displaced laterally in patients with exstrophy (Schlegel and Gearhart, 1989). These nerves are preserved in almost all exstrophy patients because potency is preserved after surgery. However, retrograde ejaculation may occur after bladder closure and later bladder neck reconstruction. Reconstruction of the female genitalia presents a less complex problem than in the male (Fig. 124–9). The vagina is shorter than normal, hardly greater than 6 cm in depth, but of normal caliber. The vaginal orifice is frequently stenotic and displaced anteriorly, the clitoris is bifid, and the labia, mons pubis, and clitoris are divergent. The uterus enters the vagina superiorly so that the cervix is in the anterior vaginal wall. The fallopian tubes and ovaries are normal. The clitoral halves should be joined, and the two ends of the labia minora joined to make a fourchette at the time of primary closure. Vaginal dilatation or episiotomy may be required to allow satisfactory intercourse in the mature female. The defective pelvic floor may predispose mature females to the development of uterine prolapse, making uterine suspension necessary. This usually occurs after childbirth but can occur even in the nulliparous patient. When studied in a large adult female population, 10 of 56 women developed uterine prolapse at a mean age of 16 years. Six patients had been managed with reconstruction that included a posterior iliac osteotomy (Mathews et al, 2003). Mean age at the time of osteotomy was 2.1 years. The use of the modern combined anterior innominate and vertical iliac osteotomy early in life may lead to reduction in the incidence of later prolapse. The size, distensibility, and neuromuscular function of the exstrophied bladder, as well as the size of the triangular fascial defect to which the bladder muscles attach, affect the decision to attempt repair. In the past several years, multiple basic science studies that further delineate the exact nature of the exstrophied bladder in the newborn have been published. One of the first papers to characterize the neuromuscular function of the bladder was published by Shapiro and colleagues (1985). In their work, muscarinic cholinergic receptor density and binding affinity were measured in control subjects and in patients with classic bladder exstrophy. The density of the muscarinic cholinergic receptors in both the control and exstrophy groups was similar, as was the binding affinity of the muscarinic receptor. Therefore it was thought by the authors that the neurophysiologic composition of the exstrophied bladder is not grossly altered during its anomalous development. Studies have investigated both the neural innervation of the newborn exstrophy bladder and its muscle and collagen content. Lee and colleagues (1996) looked at bladder biopsies obtained from 12 newborns with bladder exstrophy, compared with age-matched controls, and found an increase in the ratio of collagen to smooth muscle in the newborns with bladder exstrophy. In addition, using anticollagen antibodies, they evaluated various types of collagen in these bladders. Compared with normal control bladders, there was no statistical difference in the amount of type I collagen in the bladders of newborns with exstrophy at initial closure, but there was a threefold increase in type III collagen. Peppas and colleagues (1999) studied patients who gained adequate bladder capacities and were awaiting bladder neck reconstruction and found that the ratio of collagen to smooth muscle decreased markedly after a successful closure and infection-free follow-up. Lais and colleagues (1996) reported similar findings, but they measured the ratio of smooth muscle to collagen and found it increased after a successful closure. In an extension of the studies just cited, Mathews and colleagues (1999b) looked at the number of myelinated nerves per field in the newborn bladders of normal subjects and those with exstrophy. The average number of myelinated nerves per field was reduced in the exstrophy bladders compared with controls, and the difference was statistically significant. This reduction in nerve fibers appears to be the result of a lack of small fibers with preservation of larger nerve fibers. In light of the findings already mentioned, it is believed that bladder exstrophy in a newborn probably represents an earlier stage in bladder development. In a large study by Rosch and colleagues (1997) from Germany, multiple immunocytochemical and histochemical markers were examined in patients with epispadias or classic bladder exstrophy. These studies involved indirect immunocytochemistry for vasoactive intestinal polypeptide (VIP), neuropeptide Y (NPY), substance P (SP), calcitonin gene–related product (CGRP), protein gene product (PGP) 9.5, and nicotinamide adenine dinucleotide phosphate diaphorase (NADPHd). No evidence of bladder muscle dysinnervation was found morphologically in any cases of classic bladder exstrophy. Cases of bladder exstrophy after failed reconstruction had muscle innervation deficiencies that increased subepithelial and intraepithelial innervation. Therefore although a newborn with bladder exstrophy may have a maturational delay in bladder development, these bladders have the potential for normal development after a successful initial closure. When the bladder is small, fibrosed, inelastic, and covered with polyps, functional repair may be impossible (Fig. 124–10). Novak and colleagues (2005) investigated the pathology and malignant potential of the polyps found in these small bladders. Two types of polyps were observed, with some overlap in findings: fibrotic and edematous. Both were associated with overlying squamous metaplasia in approximately 50% of cases. Varying degrees of von Brunn nests, cystitis cystica, and cystitis glandularis were noted. Cystitis glandularis was noted in a higher percentage of secondary closures. Because of the potential risk of adenocarcinoma associated with cystitis glandularis, future surveillance of these patients with urine cytology and cystoscopy as they enter adulthood is recommended. The more normal bladder may be invaginated, or it may bulge through a small fascial defect, indicating the potential for satisfactory capacity after successful initial closure. Also, not until examination under anesthesia can the true defect be adequately evaluated because bladders that appear small in the nursery may have a good bit of bladder sequestered below the fascial defect. The depth of this extension often cannot be appreciated unless the infant is totally relaxed under anesthesia. Bladder function was assessed in a group of continent exstrophy patients with normal reflexive bladders. Normal cystometrograms were obtained in 70% to 90% of cases (Toguri et al, 1987). Diamond and colleagues (1999), looking at 30 patients with bladder exstrophy at various stages of reconstruction, found that 80% of patients had compliant and stable bladders before bladder neck reconstruction. After bladder neck reconstruction, approximately half of the patients maintained normal bladder compliance and a lesser number maintained normal stability. The authors believed that compliance and stability were impaired after bladder neck reconstruction and that 25% of patients with exstrophy may maintain normal detrusor function after reconstruction. In an earlier paper by Hollowell and colleagues (1993), 13 of 21 children revealed involuntary contractions and only 4 revealed stable bladders before bladder neck reconstruction. Also, 7 of 21 had increased pressures (>10 cm H2O), suggesting decreased compliance. The difference in findings between these two urodynamic studies is difficult to explain from an experimental perspective. However, standardized methods of bladder neck repair do not exist, and these differences may be reflected in the different urodynamic findings after bladder neck repair in these two groups of patients. Several interesting aspects of the microstructure of the bladder in children with bladder exstrophy were noted by Mathews and colleagues (2004) using specimens obtained from children with bladder exstrophy at various stages of reconstruction (newborn bladder closure, bladder neck reconstruction, augmentation cystoplasty). At the cellular level, important differences were noted. Caveoli, which are important intracellular structures involved in cell-cell signaling, were found to be normal in the patients with a successful closure and improvement in bladder capacity and significantly lacking in the patients who required eventual augmentation cystoplasty (Fig. 124–11). In addition, the ultrastructure of cells in the patients in whom closure failed was noted to be abnormal. (From Matthews RI, Wills M, Perlman E, et al. Neural innervation of the newborn exstrophy bladder: an immunohistological study. J Urol 1999;162:506.) Exstrophy complex includes a spectrum of anomalies ranging from epispadias to cloacal exstrophy. Many variations in anatomy and types of defects have been noted. Because there is probably a common embryologic origin for all of these defects, they all share many or some of the defects noted in the three major components of the complex—skeletal, urinary, and genital. The presence of a characteristic musculoskeletal defect of the exstrophy anomaly with no major defect in the urinary tract has been named “pseudoexstrophy” (Marshall and Muecke, 1968). Predominant characteristics include an elongated, low-set umbilicus and divergent rectus muscles that attach to the separated pubic bones (Fig. 124–12). In this variant, the mesodermal migration has been interrupted in its superior aspect only, thus wedging apart the musculoskeletal elements of the lower abdominal wall without obstructing the formation of the genital tubercle. In the superior vesical fissure variant of the exstrophy complex, the musculature and skeletal defects are exactly the same as those in classic exstrophy; however, the persistent cloacal membrane ruptures only at the uppermost portion, and a superior vesical fistula that actually resembles a vesicostomy results. Bladder extrusion is minimal and is present only over the normal umbilicus (see Fig. 124–1B). In a large review of an exstrophy database of more than 815 patients, Lowentritt and colleagues (2005) reported 25 exstrophy complex variants, 6 of which were cloacal exstrophy variants. Continence rates after bladder neck repair were compatible with classic exstrophy (Fig. 124–13). Three cases were reported by Arap and Giron (1986) in which the patients had classic musculoskeletal defects, and two of the three were continent. Of the two male patients, one had an associated complete epispadias and the other had a completely normal penis. Therefore the external genital manifestations in duplicate exstrophy can be quite variable. In addition to pseudoexstrophy, superior vesical fissure, and duplicated exstrophy, isolated occurrences of a fourth entity, “covered exstrophy,” have been reported (Cerniglia et al, 1989). This has also been referred to as split symphysis variant. A common factor in these patients is the presence of musculoskeletal defect associated with classic exstrophy but no significant defect of the urinary tract. Chandra and colleagues (1999) reported a covered exstrophy with incomplete duplication of the bladder. However, in most cases of covered exstrophy (Narasimharao et al, 1985; Cerniglia et al, 1989), there has been an isolated ectopic bowel segment present on the inferior abdominal wall near the genital area, which can be either colon or ileum with no connection with the underlying gastrointestinal tract and only epispadias in the male. A patient seen at our institution had the standard appearance of most split symphysis variants, and one could actually see the bladder through a thin membrane of lower abdominal skin. Although all of the classic musculoskeletal defects of exstrophy were present, there was no isolated ectopic bowel segment present on the lower abdominal wall (Fig. 124–14). These patients should all undergo formal exstrophy closure at birth. There are two different forms of duplication, anteroposterior duplication and side-by-side duplication. The first form is considered a duplicate exstrophy with a patch of everted bladder mucosa on the anterior abdominal wall with a second bladder lying in the pelvis. The ureters attach to the closed bladder, rendering the superficial mucosa dry (see Fig. 124–13). The mainstay of treatment has been resection of the ectopic mucosa and closure of the abdominal wall defect. The other form of duplication involves patients who have two separately formed bladder halves in a left-right orientation with a midline septum between the bladders containing muscle. Each bladder has its own ureter and an intact sphincter. Oftentimes there is diastasis of the pubis and rectus muscle. Of six cases reported by Lowentritt and colleagues (2005) with cloacal variants, five were covered by skin and one involved duplication of the bowel and hemibladders. Cases of cloacal exstrophy are challenging in their surgical management, and the variations of the complex add to the difficulties of initial diagnosis and management. Only by using a combination of genitograms, retrograde ureterograms, and bowel continence studies was it possible to understand the complex anatomy of the patients in these groups. Interestingly, three patients had no spinal abnormalities and one had spina bifida occulta. Three patients in this group had innervation of the pelvic floor, enabling successful Pena procedures in two, with the third patient awaiting the procedure. The epispadias-bladder/cloacal exstrophy complex has a wide range of presentations. Variants are rare, but it is important to recognize the different appearances at birth because the initial treatment will greatly influence the long-term outcome. Two patient groups had better outcomes than those with classic presentations—superior vesical fissure and skin-covered cloacal exstrophy. Because the sphincter is intact, patients with superior vesical fissure went through regular toilet training and became continent without the need for a later bladder neck procedure. Many reports of exstrophy variants include these patients, leading to the wrong belief that all patients with variants perform better than those with bladder exstrophy. Lowentritt (2005) suggests that other than superior vesical fissure, all variants should be managed with formal exstrophy closure at birth and followed in the same manner as their classic presentations. Duplicated organs can be useful in later reconstruction. Cloacal variants have a lower incidence of spinal abnormalities and a higher rate of fecal continence compared with their classic presentations. Overall, these patients had a low incidence of severe comorbid conditions. Therefore appropriate treatment of these genitourinary malformations can significantly impact and improve the quality of life. Currently, the prenatal diagnosis of bladder exstrophy is difficult to delineate. Often, a diagnosis of omphalocele or gastroschisis is made and the exstrophy condition is overlooked. Ultrasound evaluation of the fetus, by means of high-resolution real-time units, allows a thorough survey of the fetal anatomy, even during routine obstetric ultrasound examinations (Gearhart et al, 1995a). Several groups have outlined important criteria for the diagnosis of classic bladder exstrophy prenatally. In these reviews, the absence of a normal fluid-filled bladder on repeated examinations suggested the diagnosis, as did a mass of echogenic tissue on the lower abdominal wall (Mirk et al, 1986; Verco et al, 1986). In a review of 25 prenatal ultrasound examinations with the subsequent birth of a newborn with classic bladder exstrophy (Gearhart et al, 1995a), several observations were made: (1) absence of bladder filling, (2) a low-set umbilicus, (3) widening pubis ramus, (4) diminutive genitalia, and (5) a lower abdominal mass that increases in size as the pregnancy progresses and as the intra-abdominal viscera increases in size (Fig. 124–15). Sweetser and colleagues (1952) initially described a staged surgical approach for bladder exstrophy. Four to 6 days before bladder closure, bilateral iliac osteotomies were performed. Epispadias repair was performed as a separate procedure. The continence procedure was limited to freeing the fibers from the intrasymphyseal band and wrapping this band around the urethra at the time of closure to increase outlet resistance. The initial staged approach to functional bladder closure included three separate stages: bladder, abdominal wall, and posterior urethral closure; bladder neck reconstruction and antireflux procedure; and later epispadias repair. This approach was recommended for most cases of exstrophy reconstruction beginning in the early 1970s (Cendron, 1971; Jeffs et al, 1972; Williams and Keaton, 1973; Gearhart and Jeffs, 1989b). Although this procedure was successful, it has been significantly modified to include bladder, abdominal wall closure, and posterior urethral closure well onto the penis in the newborn period with bilateral innominate and vertical iliac osteotomy, if indicated; epispadias repair at 6 months to 1 year of age; and bladder neck reconstruction along with antireflux procedure at age 4 to 5 years, when the child has achieved an adequate bladder capacity for bladder neck reconstruction and is motivated to participate in a postoperative voiding program (Gearhart and Jeffs, 1998). Other methods of treatment of the newborn with bladder exstrophy have been offered. Grady and Mitchell (1999) proposed combining bladder exstrophy closure with epispadias repair in the newborn period. Baka-Jakubiak (2000) recommended newborn exstrophy closure alone and combined bladder neck reconstruction and epispadias repair when the child reaches a satisfactory age for participation in a voiding program. Kelly (1995) and colleagues have recommended a staged repair in which no osteotomy is used and supplanted by a second-stage “radical soft tissue mobilization” before later urethral repair. Schrott and colleagues (1984) recommended bladder closure, ureteral reimplantation, epispadias repair, and bladder neck reconstruction in the newborn period. Lastly, Stein and colleagues (1999) recommended ureterosigmoidostomy in the newborn period with abdominal wall and bladder closure. This chapter offers a comprehensive look at all of these repairs, their outcomes, and most importantly overall complications and those specific to the different types of repairs. At birth, although the bladder mucosa is usually smooth, pink, and intact, it is also sensitive and easily denuded. In the delivery room the umbilical cord should be tied with 2-0 silk close to the abdominal wall so that the umbilical clamp does not traumatize the delicate mucosa and cause excoriation of the bladder surface. The bladder can then be covered with a nonadherent film of plastic wrap (e.g., Saran Wrap) (Fig. 124–16) to prevent sticking of the bladder mucosa to clothing or diapers. In addition, each time the diaper is changed the plastic wrap should be removed, the bladder surface irrigated with sterile saline, and clean plastic wrap placed over the bladder surface area. Successful treatment of exstrophy with functional closure demands that the potential for success in each child be carefully considered at birth. The size and functional capacity of the detrusor muscle are important considerations for the eventual success of functional closure. Correlation between apparent bladder size and the potential bladder capacity must not be confused. In minor grades of exstrophy that approach the condition of complete epispadias with incontinence, the bladder may be small yet may demonstrate acceptable capacity, either by bulging when the baby cries or by indenting easily when touched by a sterile gloved finger in the operating room with the child under anesthesia. Sometimes a good bit of previously unappreciated bladder can be discovered behind the fascia under examination with anesthesia (Gearhart and Jeffs, 1998). Once the bladder is relieved of surface irritation and repeated trauma, the small bladder can enlarge and increase in capacity with the absence of sphincter activity and with minimal outlet resistance. The exstrophied bladder that is estimated at the time of birth to have a capacity of 5 mL or more and demonstrates elasticity and contractility can be expected to develop useful size and capacity after successful bladder, posterior urethral, and abdominal wall closure with early epispadias repair (Gearhart and Jeffs, 1998). A small, fibrotic bladder patch that is stretched between the edges of the small triangular fascial defect without elasticity or contractility cannot be selected for the usual closure procedure (Gearhart and Jeffs, 1998) (see Fig. 124–10). Examination with the patient under anesthesia may at times be required to assess the bladder adequately, particularly if considerable edema, excoriation, and polyp formation have developed between birth and the time of assessment. Decisions regarding the suitability of bladder closure or the need for waiting should be made only by surgeons with a great deal of experience in the exstrophy condition (Gearhart and Jeffs, 1998). Neonatal closure, even when the bladder is small, can allow assessment of bladder potential, provides an initial step in genital reconstruction, and is helpful in reassuring the family. Some conditions preclude primary closure, including penoscrotal duplication, ectopic bowel within the extruded bladder (a relative contraindication), a hypoplastic bladder, and significant bilateral hydronephrosis. In a recent review by Lakshmanan and colleagues (2008) of cases at our institution, it was found on initial judgment that the bladder was too small for closure in 34 patients evaluated at birth. There were 27 males and 6 females who underwent delayed closure at a mean age of 13.2 months. Osteotomy was performed on 29 (85%). All had a successful delayed primary closure. Eleven (41%) of the boys had a simultaneous epispadias repair. Sixty one percent developed sufficient capacity for bladder neck reconstruction and 39% are continent. Compared with newer data by Novak and colleagues (2010), these rates are over double the continence rates seen in bladder neck repair after failed primary closure and successful secondary closure. Pelvic osteotomy performed at the time of initial closure confers several advantages including (1) easy approximation of the symphysis with diminished tension on the abdominal wall closure and elimination of the need for fascial flaps; (2) placement of the posterior vesicourethral unit deep within the pelvic ring, enhancing bladder outlet resistance; and (3) bringing the large pelvic floor muscles near the midline, where they can support the bladder neck and aid in eventual urinary control (Fig. 124–17). After pubic approximation with osteotomy, some patients show the ability to stop and start the urinary stream, experience dry intervals, and in some cases become completely continent (Gearhart and Jeffs, 1991a). In a review of a large number of patients referred to our institution after failed exstrophy procedures, it was found that a majority of the patients who had partial or complete dehiscence of the bladder or major bladder prolapse had not undergone a prior osteotomy at the time of initial bladder closure (Gearhart et al, 1993b). The authors recommend performing bilateral transverse innominate and vertical iliac osteotomy when bladder closure is performed after 72 hours of age (Fig. 124–18). In addition, if the pelvis is not malleable or if the pubic bones are more than 4 cm apart at the time of initial examination under anesthesia, osteotomy should be performed, even if closure is done before 72 hours of age. A well-coordinated surgery and anesthesia team can perform osteotomy and proceed to bladder closure without undue loss of blood or risk of prolonged anesthesia in the child. However, it must be realized that osteotomy together with posterior urethral and bladder closure and abdominal wall closure is a 5- to 7-hour procedure in these infants.
Exstrophy-Epispadias Complex
Historical Aspects
Incidence and Inheritance
Embryology
Classic Bladder Exstrophy
Anatomic Considerations
Skeletal Defects
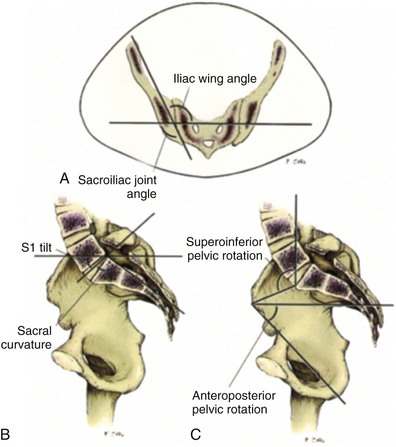
Pelvic Floor Defects
Abdominal Wall Defects
Anorectal Defects
Male Genital Defect
Female Genital Defects
Urinary Defects
Exstrophy Complex and Variants
Cloacal Exstrophy Variants—Covered Cloacal Exstrophy and Duplicate Bladder
Prenatal Diagnosis
Surgical Reconstruction of Bladder Exstrophy
Evaluation and Management at Birth
Selection of Patients for Immediate Closure
Small Exstrophy Bladder Unsuitable for Newborn Closure
Osteotomy
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Exstrophy-Epispadias Complex