A. Mark Richards, M. Gary Nicholls
Endocrine Causes of Hypertension
The true incidence and prevalence of hypertension with endocrine etiology are unknown. However, primary aldosteronism may be present in up to 12% of patients with newly diagnosed hypertension, and overall, an endocrine contribution to high blood pressure (BP) may be present in more than 12% of cases. Endocrine hypertension often remains undiagnosed because physicians consider it rare and therefore unlikely, and access to the required specialized tests is limited.
Endocrine hypertension may occur in the absence of readily observed signs and symptoms or abnormalities in routine biochemical tests. However, certain features should trigger a simple screen (Fig. 41-1), such as a family history of inherited conditions, including pheochromocytoma, neurofibromatosis, multiple endocrine neoplasia, and aldosteronism. Resistant hypertension (i.e., BP resistant to administration of three antihypertensive drugs of different classes) mandates consideration of secondary hypertension once iatrogenic factors and noncompliance have been ruled out. The differential diagnoses for endocrine hypertension will include renal parenchymal disease and renovascular disease.
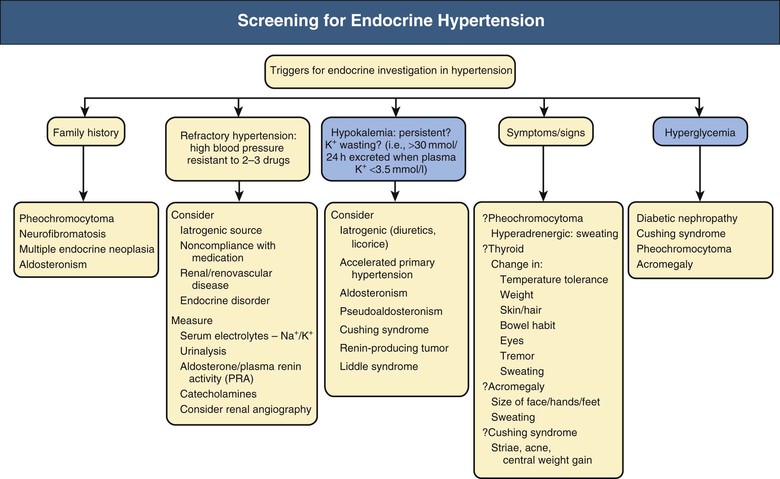
Symptoms and signs may be present. Severe hypokalemia is rare but when present may cause weakness, polyuria, and cardiac arrhythmia. Hyperadrenergic symptoms occur with pheochromocytoma. Changes in temperature tolerance, body weight, hair and skin condition, and bowel habit are clues in thyroid dysfunction or hypercortisolism. Thyroid disease, Cushing syndrome, and acromegaly are associated with typical changes in body habitus. Abnormal sweating occurs in pheochromocytoma, thyrotoxicosis, and acromegaly.
Hypokalemia and diabetes mellitus are hallmarks of endocrine pathologic processes in hypertension. Hypokalemia in the absence of diuretic use should suggest diagnoses such as primary aldosteronism, pseudoaldosteronism, renin-secreting tumor, Cushing syndrome, and accelerated hypertension of any cause. Therefore, concurrent high BP and hypokalemia should prompt consideration of endocrine hypertension. Hyperglycemia is common to Cushing syndrome, pheochromocytoma (especially when epinephrine levels are increased), acromegaly, and primary aldosteronism. Diagnosis of endocrine forms of hypertension offers the chance of cure, of altering an otherwise catastrophic natural history (e.g., in pheochromocytoma), and of applying specific and effective therapies that ameliorate other elements of the disease beyond BP control. Chapter 40 discusses aldosteronism; this chapter describes other types of endocrine hypertension.
Cushing Syndrome
Definition
This syndrome of sustained glucocorticoid excess is most often secondary to production of adrenocorticotropic hormone (ACTH) by a pituitary adenoma (Cushing disease). Less frequently, Cushing syndrome is the result of cortisol overproduction from an adrenal adenoma or carcinoma, and rarely it may be secondary to ectopic ACTH (corticotropin) secretion.1 It can also result from exogenous corticosteroid administration. The incidence of endogenous Cushing syndrome is 5 to 10 cases per 1 million population per year. Cushing disease and cortisol-secreting adrenal tumors are four times more common in women than in men. Approximately 0.5% of patients with bronchogenic carcinoma (more common in men than women) develop ectopic ACTH syndrome.
Etiology, Pathogenesis, and Epidemiology
Hypertension is present in 80% of patients with Cushing syndrome (less often when caused by exogenous synthetic corticosteroid administration) and results from an increase in both cardiac output and total peripheral resistance. The mechanisms underlying these hemodynamic changes are complex.2
In some patients, Cushing syndrome may be caused by concurrent overproduction of mineralocorticoids such as aldosterone, 11-deoxycorticosterone, and corticosterone. Although cortisol is capable of binding the mineralocorticoid receptor (MR), usually it does not because of the renal enzyme 11β-hydroxysteroid dehydrogenase type 2 (β-HSD2), which inactivates cortisol to corticosterone, thereby preventing its binding to the MR. However, in the patient with low β-HSD2 activity or extremely high cortisol levels (e.g., ectopic ACTH syndrome), there may be sufficient excess cortisol that MR binding occurs. Furthermore, alterations in oxidation-reduction (redox) status, potentially triggered by metabolic disturbance and cytokine activation, may alter the effects of corticosteroids at the MR function, converting cortisol from its receptor antagonist role seen in health to an agonist in inflammatory states.3
Subsequent cardiac and vascular MR activation may foster adverse cardiovascular inflammatory, hypertrophic, and fibrotic changes. Inhibition of the vasodilator nitric oxide (NO) by cortisol may also contribute to the hypertension, along with enhanced pressor responsiveness to catecholamines and angiotensin II (Ang II), heightened cardiac inotropic sensitivity to β-adrenergic stimulation, and increased plasma volume.2 If affected, the sympathetic nervous system (SNS) and renin-angiotensin system (RAS) are suppressed, even though circulating levels of renin substrate are increased. Some have proposed that elevated levels of vasoconstrictor hormones (vasopressin, endothelin-1, thromboxane, insulin) or subnormal concentrations of vasodilator systems (kallikrein-kinins, prostaglandins) contribute to the hypertension of Cushing syndrome, but the evidence is weak.2 The vasodilator atrial natriuretic peptide (ANP) is elevated in Cushing syndrome, although biologic responses to ANP are impaired; in theory, ANP resistance could also contribute to the hypertension.4 Adipokines, including leptin and resistin, plus release of proinflammatory cytokines (tumor necrosis factor α, interleukin-6) may also contribute to the increased cardiovascular risk observed in Cushing syndrome.5
Successful treatment of Cushing disease or removal of an underlying adrenal adenoma usually results in BP reduction and partial return of the previously impaired nocturnal fall in arterial pressure, although hypertension persists in a sizable minority of patients.
Clinical Manifestations
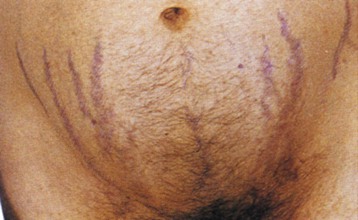
Clinical features in Cushing disease result from elevated circulating levels of pro-opiomelanocortin hormones, including ACTH (increased pigmentation) and cortisol (central adiposity, insulin resistance or diabetes, muscle wasting/weakness, plethoric facies, purple striae [Fig. 41-2], easy bruising, osteoporosis, psychological problems). In some patients, androgen effects are observed (hirsutism, acne, virilization) and may be striking in those with adrenal adenoma or carcinoma. Ectopic ACTH syndrome caused by small cell bronchial carcinoma or other tumors (e.g., bronchial or thymic carcinoid) presents typically as a wasting disease, often with hyperpigmentation and hypokalemia. Hypertension is often associated with left ventricular hypertrophy, which can be disproportionate to BP, and frank cardiac failure is occasionally the presenting feature.6
Differential Diagnosis
Pseudo–Cushing syndrome can occur with a sustained high intake of alcohol by inducing augmented cortisol secretion and reduced cortisol metabolism caused by hepatic damage. Routine diagnostic tests are unable to distinguish alcoholic pseudo–Cushing syndrome from true Cushing syndrome, and reassessment after alcohol withdrawal may be required.7
Depression is associated with increased cortisol level. In obesity, the plasma clearance rate of cortisol is increased, leading to a slightly elevated production rate of cortisol, although plasma levels are normal. Careful psychological and physical screening will usually differentiate these patients from those with Cushing syndrome.
Diagnosis
An elevated urinary free cortisol excretion (or early-morning cortisol-creatinine ratio) and absence of suppression of 8 am plasma cortisol after a low dose of dexamethasone (1 mg at midnight) are the usual initial tests. Screening may also incorporate checks for elevated late-night salivary cortisol.
Further key investigations include computed tomography (CT) and magnetic resonance imaging (MRI) of the pituitary and adrenal glands and plasma ACTH (suppressed in cortisol-secreting adrenal tumors but elevated in ectopic ACTH syndrome). Further tests will depend on findings at this point. A high-dose dexamethasone suppression test partially suppresses ACTH in patients with pituitary tumors but not with ectopic ACTH. The response of plasma ACTH to a dose of corticotropin-releasing hormone may also help distinguish pituitary from adrenal or ectopic tumors. Simultaneous bilateral inferior petrosal sinus sampling for ACTH measurements is useful in differentiating Cushing disease from ectopic ACTH syndrome, when the previous tests give equivocal results. Imaging of the thorax, abdomen, and pelvis is indicated when ACTH-producing carcinoid tumors are suspected.
Treatment and Prognosis
Untreated, patients with Cushing syndrome have 50% 5-year mortality because of cardiovascular risk from hypertension, along with glucose intolerance and insulin resistance, hyperlipidemia, obesity, and elevated fibrinogen levels.8
Cure rates in treatment of Cushing disease are 80% to 90% by selective removal of a pituitary microadenoma and 50% for pituitary macroadenomas. Cushing syndrome caused by adrenal adenoma is almost always cured by unilateral adrenalectomy. However, in adrenal carcinoma, median survival is less than 2 years. The prognosis is also poor when Cushing syndrome results from ectopic ACTH syndrome caused by small cell bronchial carcinoma. If the ACTH-producing tumor is benign and can be located, however, removal usually leads to cure. After cure of Cushing syndrome, approximately 30% of patients have persistent hypertension.9
In managing the hypertension from Cushing syndrome, no evidence supports the use of any antihypertensive class over another. Potassium-losing diuretics can exacerbate both hypokalemia and glucose intolerance, whereas potassium-sparing diuretics, usually in combination with other antihypertensive agents, may correct hypokalemia and reduce edema while lowering BP.
Pheochromocytoma
Definition
Pheochromocytoma has challenged and fascinated clinicians since its first description by Frankel in 1886. Because clinical manifestations can mimic a wide spectrum of other disorders, the diagnosis is frequently delayed or missed altogether, sometimes with fatal consequences.10
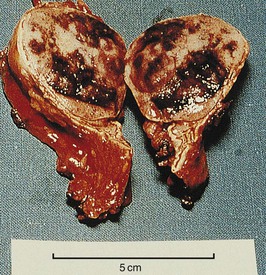
Pheochromocytoma refers to a dusky tumor whose cells stain brown with chromium salts. Such tumors arise most frequently within the adrenal glands (Fig. 41-3), but approximately 10% are extra-adrenal (paraganglioma). Although the majority of pheochromocytomas are benign, 10% metastasize to regional lymph nodes and beyond. Histologic features are not a reliable guide to malignant behavior. The tumors can secrete a wide variety of hormones but most characteristically produce norepinephrine, epinephrine, and dopamine, with patterns differing among patients. Few paragangliomas produce epinephrine. Very high dopamine production is associated with malignant disease or a large tumor mass.
Etiology, Pathogenesis, and Epidemiology
The prevalence of pheochromocytoma in patients with hypertension in general medical outpatient clinics is 0.1% to 0.6%.11 As with a firm diagnosis, however, the prevalence may be considerably higher because many people die with unsuspected pheochromocytoma.12
Pheochromocytomas can be sporadic or familial. Whereas sporadic cases are usually unicentric and unilateral, familial pheochromocytomas are often multicentric and bilateral. Familial pheochromocytomas are the result of a germ-line mutation in one of five genes: the RET gene leading to multiple endocrine neoplasia type 2; the von Hippel–Lindau (VHL) gene, which causes the von Hippel–Lindau syndrome; the neurofibromatosis type 1 (NF1) gene resulting in von Recklinghausen disease; and the genes encoding the B and D subunits of mitochondrial succinate dehydrogenase (SDHB and SDHD), which are associated with familial paragangliomas and pheochromocytomas. More recently, mutations in KIF1Bβ, TMEM127, and MAX genes have also been associated with pheochromocytoma.13
Table 41-1 outlines clinical features of syndromes associated with pheochromocytoma. For patients with apparently sporadic pheochromocytomas, an underlying germ-line mutation of the genes mentioned may be present in about 20% of cases and should be considered in younger patients (<50 years) and those with multifocal or extra-adrenal tumors.10,14 Patients found to harbor a germ-line mutation need to be identified for appropriate guidance of medical management for them and their families.
Table 41-1
Primary clinical features of syndromes associated with pheochromocytoma.
| Main Clinical Features of Syndromes Associated with Pheochromocytoma | |
| Associated Syndrome | Clinical Manifestations |
| Von Hippel–Lindau Syndrome | |
| Type 1 (no pheochromocytoma) | |
| Type 2 (with pheochromocytoma) | |
| Multiple Endocrine Neoplasia (MEN) Type 2* | |
| Type 2A (MEN-2A) | |
| Type 2B (MEN-2B) | |
| Other Syndromes | |
| Neurofibromatosis type 1 | |
| Paraganglioma syndromes | |
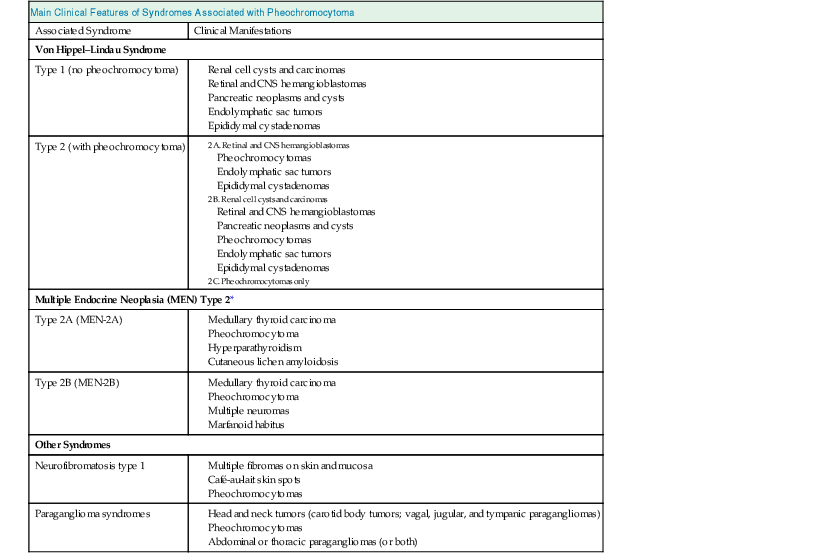
* A third type of MEN type 2 consists of familial medullary thyroid carcinoma only (without pheochromocytoma).
CNS, Central nervous system.
Clinical Manifestations
Clinical features of pheochromocytoma reflect episodic or continuous overproduction of catecholamines and depend in part on which catecholamine dominates. Symptoms include headache, sweating, palpitations, anxiety, and pallor (Fig. 41-4).15
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


