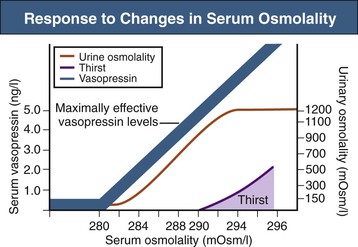
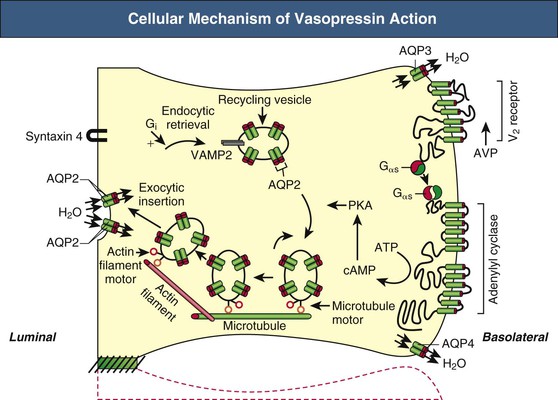
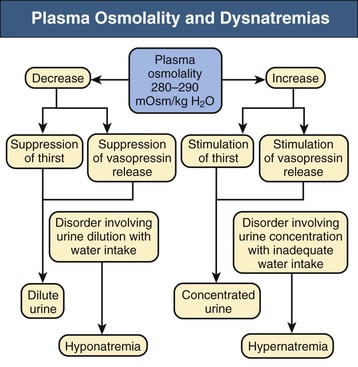
Tomas Berl, Chirag Parikh The maintenance of the tonicity of body fluids within a narrow physiologic range is made possible by homeostatic mechanisms that control the intake and excretion of water. Vasopressin, also known as arginine vasopressin (AVP) or antidiuretic hormone (ADH), governs the excretion of water by its effect on the renal collecting system. Osmoreceptors located in the hypothalamus control the secretion of vasopressin in response to changes in tonicity. In the steady state, water intake matches water losses. Water intake is regulated by the need to maintain a physiologic serum osmolality of 285 to 290 mOsm/kg. Despite major fluctuations of solute and water intake, the total solute concentration (i.e., the tonicity) of body fluids is maintained virtually constant. The ability to dilute and to concentrate the urine allows wide flexibility in urine flow (see Chapter 2). During water loading, the diluting mechanisms permit excretion of 20 to 25 liters of urine daily, and during water deprivation, the urine volume may be as low as 0.5 l/day.1,2 Vasopressin plays a critical role in determining the concentration of urine. It is a 1099-d cyclic peptide and is synthesized and secreted by the specialized supraoptic and paraventricular magnocellular nuclei in the hypothalamus. Vasopressin has a short half-life of about 15 to 20 minutes and is rapidly metabolized in the liver and the kidney. Substances restricted to the extracellular fluid (ECF), such as hypertonic saline and mannitol, decrease cell volume by acting as effective osmoles and enhancing osmotic water movement from the cell. This stimulates vasopressin release; in contrast, urea and glucose readily cross cell membranes and thus do not cause changes in cell volume. The “osmoreceptor” cells, located close to the supraoptic nuclei in the anterior hypothalamus, are sensitive to changes in plasma osmolality as small as 1% and bring about the release of vasopressin by a pathway that involves the activation of TRPV4 (transient receptor potential) channels. In humans, the osmotic threshold for vasopressin release is 280 to 290 mOsm/kg2,3 (Fig. 8-1). This system is so efficient that plasma osmolality usually does not vary by more than 1% to 2% despite wide fluctuations in water intake. There are several other nonosmotic stimuli for vasopressin secretion. Decreased effective circulating volume (e.g., heart failure, cirrhosis, vomiting) causes discharge from parasympathetic afferent nerves in the carotid sinus baroreceptors and increases vasopressin secretion. Other nonosmotic stimuli include nausea, postoperative pain, and pregnancy. Much higher vasopressin levels can be achieved with hypovolemia than with hyperosmolality, although a large (7%) decrease in blood volume is required before this response is elicited. Vasopressin binds three types of receptors coupled to G proteins: the V1a (vascular and hepatic), V1b (anterior pituitary), and V2 renal receptors. The V2 receptor is primarily localized in the collecting duct and leads to an increase in water permeability through aquaporin 2 (AQP2), which is a member of a family of cellular water transporters4 (Fig. 8-2). AQP1 is localized in the apical and basolateral region of the proximal tubule epithelial cells and the descending limb of Henle loop and accounts for the high water permeability of these nephron segments. Because AQP1 is constitutively expressed, it is not subject to regulation by vasopressin. In contrast, AQP2 is found exclusively in apical plasma membranes and intracellular vesicles in the collecting duct principal cells. Vasopressin affects both the short-term and the long-term regulation of AQP2. The short-term regulation, also described as the “shuttle hypothesis,” explains the rapid and reversible increase (within minutes) in collecting duct water permeability after vasopressin administration. This involves the insertion of water channels from subapical vesicles into the luminal membrane. Long-term regulation involves vasopressin-mediated increased transcription of genes involved in AQP2 production and occurs if circulating vasopressin levels are elevated for 24 hours or more. The maximal water permeability of the collecting duct epithelium is increased as a consequence of an increase in the total number of AQP2 channels per cell. This process is not readily reversible. Aquaporins 3 and 4 are located on the basolateral membranes of the collecting duct (Fig. 8-2) and are probably involved in water exit from the cell. AQP3 is also urea permeable and, under the stimulus of vasopressin, increases the permeability of the collecting duct to urea, resulting in its movement into the interstitium. AQP4 is also found in the hypothalamus and is a candidate osmoreceptor for the control of vasopressin release. Hypertonicity is the most potent stimulus for thirst, with a change of only 2% to 3% in plasma osmolality producing a strong desire to drink. The osmotic threshold for thirst usually occurs at 290 to 295mOsm/kg H2O and is above the threshold for vasopressin release (see Fig. 8-1). It closely approximates the level at which maximal concentration of urine is achieved. Hypovolemia, hypotension, and angiotensin II (Ang II) are also stimuli for thirst. Between the limits imposed by the osmotic thresholds for thirst and vasopressin release, plasma osmolality may be regulated more precisely by small, osmoregulated adjustments in urine flow and water intake. The exact level at which balance occurs depends on various factors, such as insensible losses through skin and lungs, the gains incurred from drinking water and eating, and water generated from metabolism. Urine volume can be considered as having two components. The osmolar clearance (Cosm) is the volume needed to excrete solutes at the concentration of solutes in plasma. The free water clearance (Cwater) is the volume of water that has been added to (positive Cwater) or subtracted from (negative Cwater) isotonic urine (Cosm) to create either hypotonic or hypertonic urine. Urine volume flow (V) comprises the isotonic portion of urine (Cosm) plus the free water clearance (Cwater). Therefore: The Cosm, solute clearance is determined by urine flow, urine osmolality, and plasma osmolality Posm as follows: Therefore: This relationship reflects the following: 1. In hypotonic urine (Uosm < Posm), Cwater is positive. 2. In isotonic urine (Uosm = Posm), Cwater is zero. 3. In hypertonic urine (Uosm > Posm), Cwater is negative (i.e., water is retained). If excretion of free water in a polyuric patient is unaccompanied by water intake, the patient will become hypernatremic. Conversely, failure to excrete free water with increased water intake can cause hyponatremia. A limitation of the previous equation is that it fails to predict clinically important alterations in plasma tonicity and serum sodium concentration (serum [Na+]) because it factors in urea. Urea is an important component of urinary osmolality; however, because it crosses cell membranes readily, urea does not establish a transcellular osmotic gradient and does not cause water movement between fluid compartments. Therefore, urea does not influence serum Na+ concentration or the release of vasopressin. As a result, changes in serum [Na+] are better predicted by electrolyte free water clearance [Cwater(e)]. The equation can be modified, replacing Posm by plasma [Na+] (PNa) and the urine osmolality by urine [Na+] and urine [K+], potassium concentration, (UNa + UK): If UNa + UK is less than PNa, then Cwater(e) is positive and serum [Na+] increases. If UNa + UK is greater than PNa, then Cwater(e) is negative and serum [Na+] decreases. In the clinical setting, it is more appropriate to use the equation for electrolyte free clearance to predict if a patient’s serum [Na+] will increase or decrease in the face of the prevailing water excretion. For example, in a patient with high urea excretion, the original equation would predict negative water excretion and a decrease in serum [Na+]; but in fact, [Na+] increases, which is accurately predicted by the latter equation. The countercurrent mechanism of the kidneys, which allows urinary concentration and dilution, acts in concert with the hypothalamic osmoreceptors through vasopressin secretion to keep serum [Na+] and tonicity within a very narrow range5 (Fig. 8-3). A defect in the urine-diluting capacity coupled with excess water intake leads to hyponatremia. A defect in urine-concentrating ability with inadequate water intake leads to hypernatremia. Serum [Na+] along with its accompanying anions accounts for nearly all the osmotic activity of the plasma. Calculated serum osmolality is given by 2[Na+] + BUN (mg/dl)/2.8 + glucose (mg/dl)/18, where BUN is blood urea nitrogen. The addition of other solutes to ECF results in an increase in measured osmolality (Table 8-1). Solutes that are permeable across cell membranes do not cause water movement and cause hypertonicity without cellular dehydration, as in uremia or ethanol intoxication. By contrast, in diabetic ketoacidosis with an increase in plasma glucose, which cannot move freely across cell membranes in the absence of insulin, water moves from the cells to the ECF, leading to cellular dehydration and lowering serum [Na+]. This can be viewed as translocational because the decrease in serum [Na+] does not reflect change in total body water but rather the movement of water from intracellular to extracellular space. A correction whereby a decrease in serum [Na+] of 1.6 mmol/l for every 100 mg/dl (5.6 mmol/l) of glucose is used, but this may somewhat underestimate the impact of glucose to decrease serum [Na+]. Pseudohyponatremia occurs when the solid phase of plasma (usually 6% to 8%) is increased by large increments in either lipids or proteins (e.g., in hypertriglyceridemia and paraproteinemias). Serum osmolality is normal in pseudohyponatremia. This false result occurs because the usual method that measures the concentration of sodium uses whole plasma and not just the liquid phase, in which the concentration of sodium is 150 mmol/l. Many laboratories are now moving to direct ion-selective potentiometry, which will give the true aqueous sodium activity. In the absence of a direct-reading potentiometer, an estimate of plasma water can be obtained from the following well-validated formula6: where L and P refer to the total lipid and protein concentration (in g/l), respectively. For example, if the formula reveals that plasma water is 90% of the plasma sample rather than the normal 93% (which yields a serum sodium concentration of 140 mmol/l as 150 × 0.93 = 140), the concentration of measured sodium would be expected to decrease to 135 mmol/l (150 × 0.90). In normal individuals, total body water is approximately 60% of body weight (50% in women and obese individuals). With hyponatremia or hypernatremia, the change in total body water can be calculated from the serum [Na+] by the following formula: where [Na+]obs is observed sodium concentration (in mmol/l) and W is body weight (in kilograms). By use of this formula, a change of 10 mmol/l in the serum [Na+] in a 70-kg individual is equivalent to a change of 3 liters in free water. Hyponatremia is defined as serum [Na+] of less than 135 mmol/l and equates with a low serum osmolarity once translocational hyponatremia and pseudohyponatremia are ruled out. True hyponatremia develops when normal urine-diluting mechanisms are disturbed7 (Fig. 8-4). Hyponatremia may result from intrarenal factors, such as a diminished glomerular filtration rate (GFR) and an increase in proximal tubular fluid and Na+ reabsorption, which decrease distal delivery of filtrate to the diluting segments of the nephron. Also, hyponatremia may result from a defect in Na+-Cl− transport out of the water-impermeable segments of the nephrons (thick ascending limb of Henle loop [TAL] or distal convoluted tubule). Most frequently, hyponatremia results from continued vasopressin secretion by nonosmotic mechanisms despite the presence of serum hypo-osmolality. Once pseudohyponatremia and translocational hyponatremia are ruled out and the patient is established as truly hypo-osmolar, the next step is to classify the patient as hypovolemic, euvolemic, or hypervolemic (Fig. 8-5). A patient with hypovolemic hyponatremia has both a total body Na+ and a water deficit, with the Na+ deficit exceeding the water deficit. This occurs in patients with high gastrointestinal and renal losses of water and solute accompanied by free water or hypotonic fluid intake. The underlying mechanism is the nonosmotic release of vasopressin stimulated by volume contraction, which maintains vasopressin secretion despite the hypotonic state. Measurement of urine [Na+] is a useful tool in helping to diagnose these conditions (Fig. 8-5). In the patient with diarrhea or vomiting, the kidney responds to volume contraction by conserving Na+ and Cl−. A similar pattern is observed in burn victims and patients with sequestration of fluids in third spaces, as in the peritoneal cavity with peritonitis or pancreatitis or in the bowel lumen with ileus. In all these patients, urine [Na+] is usually less than 10 mmol/l, and the urine is hyperosmolar. An exception to this is in patients with vomiting and metabolic alkalosis. Here, the increased bicarbonate ion (HCO3−) excretion obligates simultaneous cation excretion such that urine [Na+] may exceed 20 mmol/l despite severe volume depletion, but in this clinical setting the urine [Cl−] is lower than 10 mmol/l. Likewise, in chronic renal insufficiency, renal salt conservation is impaired and urine [Na+] may be high. Diuretic use is one of the most common causes of hypovolemic hyponatremia associated with a high urine [Na+]. Loop diuretics inhibit Na+-Cl− reabsorption in the TAL. This interferes with the generation of a hypertonic medullary interstitium. Therefore, even though volume contraction leads to increased vasopressin secretion, responsiveness to vasopressin is diminished and free water is excreted. In contrast, thiazide diuretics act in the distal tubule by interfering with urine dilution rather than with urine concentration, limiting free water excretion. Hyponatremia usually occurs within 14 days of initiation of therapy, although one third of patients present within 5 days. Underweight women and elderly patients appear to be most susceptible. Postulated mechanisms for diuretic-induced hyponatremia include the following: Water retention can mask the physical findings of hypovolemia, thereby making the patients with diuretic-induced hyponatremia appear euvolemic. A salt-losing state may occur in patients with advanced chronic renal impairment (GFR <15 ml/min), particularly from interstitial disease. It is characterized by hyponatremia and hypovolemia. In proximal type 2 renal tubular acidosis, there is renal Na+ and K+ wastage despite only moderate renal impairment, and bicarbonaturia obligates urine Na+ excretion. Mineralocorticoid deficiency is characterized by hyponatremia with ECF volume contraction, urine [Na+] above 20 mmol/l, and high serum K+, urea, and creatinine. Decreased ECF volume provides the nonosmotic stimulus for vasopressin release. An osmotically active, non-reabsorbable solute obligates the renal excretion of Na+ and results in volume depletion. In the face of continuing water intake, the diabetic patient with severe glycosuria, the patient with a urea diuresis after relief of urinary tract obstruction, and the patient with mannitol diuresis all undergo urinary losses of Na+ and water, leading to hypovolemia and hyponatremia. Urine [Na+] is typically above 20 mmol/l. The ketone bodies β-hydroxybutyrate and acetoacetate also obligate urinary electrolyte losses and aggravate the renal Na+ wasting seen in diabetic ketoacidosis, starvation, and alcoholic ketoacidosis. Cerebral salt wasting is a syndrome described primarily in patients with subarachnoid hemorrhage. The primary defect is salt wasting from the kidneys with subsequent volume contraction, which stimulates vasopressin release. The exact mechanism is not understood, but it is postulated that brain natriuretic peptide increases urine volume and Na+ excretion. The diagnosis requires evidence of inappropriate sodium losses and reduced effective blood volume. These criteria are rarely fulfilled, suggesting that cerebral salt wasting is overdiagnosed.8 In hypervolemia, if the total body water is increased more than total body Na+ hyponatremia occurs, as in congestive heart failure (CHF), nephrotic syndrome, and cirrhosis, all of which are associated with impaired water excretion (see Fig. 8-5 and Chapter 7). Edematous patients with CHF have reduced effective intravascular volume as a result of decreased systemic mean arterial pressure and cardiac output. This reduction is sensed by aortic and carotid baroreceptors activating nonosmotic pathways, and vasopressin is released. In addition, the relative “hypovolemic” state stimulates the renin-angiotensin axis and increases norepinephrine production, which in turn decreases GFR. The decrease in GFR leads to an increase in proximal tubular reabsorption and a decrease in water delivery to the distal tubule. The neurohumorally mediated decrease in delivery of tubular fluid to the distal nephron and an increase in vasopressin secretion mediate hyponatremia by limiting Na+-Cl− and water excretion. In addition, low cardiac output and high Ang II levels are potent stimuli of thirst. There is also excessive intracellular targeting of AQP2 to the apical cell membrane of the collecting duct (Fig. 8-6).9 These effects most likely result from high circulating levels of vasopressin. As cardiac function improves with afterload reduction, plasma vasopressin decreases, with concomitant improvement in water excretion. The degree of hyponatremia has also been correlated with the severity of cardiac disease and with patient survival; serum [Na+] of less than 125 mmol/l reflects severe CHF. Patients with cirrhosis and hepatic insufficiency also have increased ECF volume (ascites, edema). Because of splanchnic venous dilation, these patients have increased plasma volume. Unlike patients with CHF, cirrhotic patients have an increased cardiac output because of multiple arteriovenous fistulas in their alimentary tract and skin. Vasodilation and arteriovenous fistulas lead to a decrease in mean arterial blood pressure. As the severity of cirrhosis increases, there are progressive increases in plasma renin, norepinephrine, vasopressin, and endothelin. There is also an associated decline in mean arterial pressure and serum [Na+]. In experimental models of cirrhosis, there is increased expression of vasopressin-regulated AQP2 in collecting ducts.4 In some patients with nephrotic syndrome, especially children with minimal change disease, hypoalbuminemia and lowered plasma oncotic pressure alter Starling forces, leading to intravascular volume contraction. Most patients with nephrotic syndrome appear to have a renal defect in sodium excretion resulting in increased effective circulating volume. In experimental models of nephrotic syndrome, expression of AQP2 and AQP3 in the renal collecting ducts is downregulated.4 Patients with advanced renal impairment, either acute or chronic, have a profound increase in fractional excretion of Na+ to maintain normal salt balance given the overall decreased number of functioning nephrons. Edema usually develops when the Na+ ingested exceeds the kidneys’ capacity to excrete this load. Likewise, if water intake exceeds threshold, there is positive water balance and hyponatremia. At a GFR of 5 ml/min, only 7.2 liters of filtrate is formed daily. Approximately 30%, or 2.2 liters, of this filtered fluid will reach the diluting segment of the nephron, which is therefore the maximum solute-free water that can be excreted daily. Euvolemic hyponatremia is the most common dysnatremia in hospitalized patients. In these patients, no physical signs of increased total body Na+ are detected. Glucocorticoid deficiency causes impaired water excretion in patients with primary and secondary adrenal insufficiency. Elevation of vasopressin accompanies the water excretion defect resulting from anterior pituitary and adrenocorticotropic hormone (ACTH, corticotropin) deficiency. This can be corrected by physiologic doses of corticosteroids. In addition, implicated vasopressin-independent factors are impaired renal hemodynamics and decreased distal fluid delivery to the diluting segments of the nephron.
Disorders of Water Metabolism
Physiology of Water Balance
Vasopressin
Osmotic Stimuli for Vasopressin Release
Nonosmotic Stimuli for Vasopressin Release
Mechanism of Vasopressin Action
Thirst and Water Balance
Quantitation of Renal Water Excretion
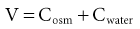
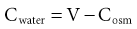
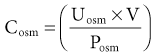
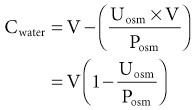
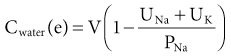
Serum Sodium Concentration, Osmolality, and Tonicity
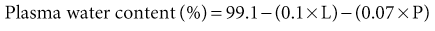
Estimation of Total Body Water
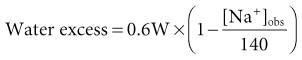
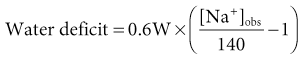
Hyponatremic Disorders
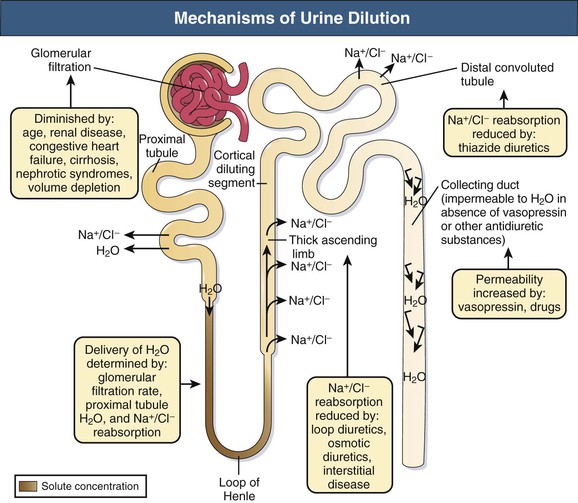
Etiology and Classification of Hyponatremia
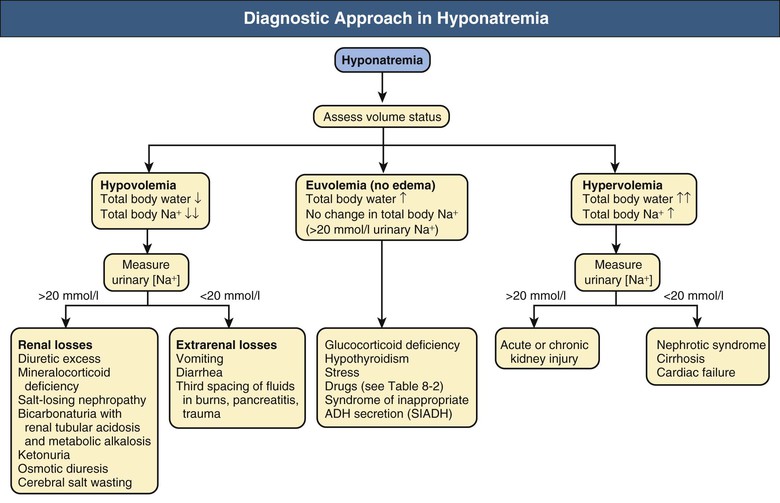
Hypovolemia: Hyponatremia Associated with Decreased Total Body Sodium
Gastrointestinal and Third-Space Sequestered Losses
Diuretics
Salt-Losing Nephropathy
Mineralocorticoid Deficiency
Osmotic Diuresis
Cerebral Salt Wasting
Hypervolemia: Hyponatremia Associated with Increased Total Body Sodium
Congestive Heart Failure
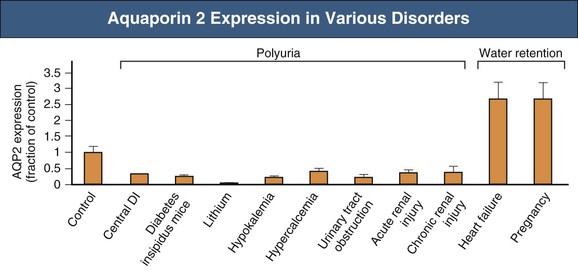
Hepatic Failure
Nephrotic Syndrome
Advanced Chronic Renal Impairment
Euvolemia: Hyponatremia Associated with Normal Total Body Sodium
Glucocorticoid Deficiency
Hypothyroidism
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree