I. David Weiner, Stuart L. Linas, Charles S. Wingo
Disorders of Potassium Metabolism
Potassium disorders are some of the most frequently encountered fluid and electrolyte abnormalities in clinical medicine. Patients with disorders of potassium metabolism may be asymptomatic or they may have symptoms ranging from mild weakness to sudden death. When the serum potassium level is verified as abnormal, correction is essential, but inappropriate treatment can worsen symptoms and even lead to death.
Normal Physiology of Potassium Metabolism
Potassium Intake
Potassium is essential for many cellular functions, is present in most foods, and is excreted primarily by the kidney. The typical Western diet contains about 70 to 150 mmol of potassium daily. The gastrointestinal (GI) tract efficiently absorbs potassium, and total dietary potassium intake varies the composition of the diet. Table 9-1 shows the potassium content of several foods high in potassium.1
Table 9-1
Amount of potassium (K+) in select foods with high potassium content.
| Potassium Content of Select High-Potassium Foods | |||||
| Food | Common Serving Size | Serving Size (g) | K+ per Serving (mg) | K+ per Serving (mmol) | mmol K+/100 g |
| Apricots, dried | 10 halves | 35 | 407 | 10 | 30 |
| Banana | 1 banana | 118 | 422 | 11 | 9 |
| Cabbage, Chinese (pak-choi) | 1 cup | 170 | 631 | 16 | 10 |
| Chickpeas or garbanzo beans | 1 cup | 164 | 477 | 12 | 7 |
| Chocolate, semisweet | 1 cup | 168 | 613 | 16 | 9 |
| Dates | ~5 dates | 42 | 272 | 7 | 17 |
| French fried potato | 1 large | 169 | 979 | 25 | 15 |
| Lentils, boiled without salt | 1 cup | 198 | 731 | 19 | 9 |
| Lima beans, boiled without salt | 1 cup | 188 | 955 | 24 | 13 |
| Papaya | 1 papaya | 304 | 553 | 14 | 5 |
| Plantains, raw | 1 medium | 179 | 893 | 23 | 13 |
| Potatoes, baked | 1 medium potato | 202 | 1081 | 28 | 14 |
| Raisins, seedless | 1 cup | 145 | 1086 | 28 | 19 |
| Refried beans, traditional style | 1 cup | 252 | 847 | 22 | 9 |
| Sauce, pasta, spaghetti/marinara, ready-to-serve | 1 cup | 250 | 798 | 20 | 8 |
| Soybeans, green, cooked, boiled, drained | 1 cup | 180 | 970 | 25 | 14 |
| Sweet potato, canned | 1 cup | 255 | 796 | 20 | 8 |
| Tomato sauce, canned | 1 cup | 245 | 811 | 21 | 8 |
| Yogurt, plain, low fat | 8 oz | 227 | 579 | 15 | 7 |
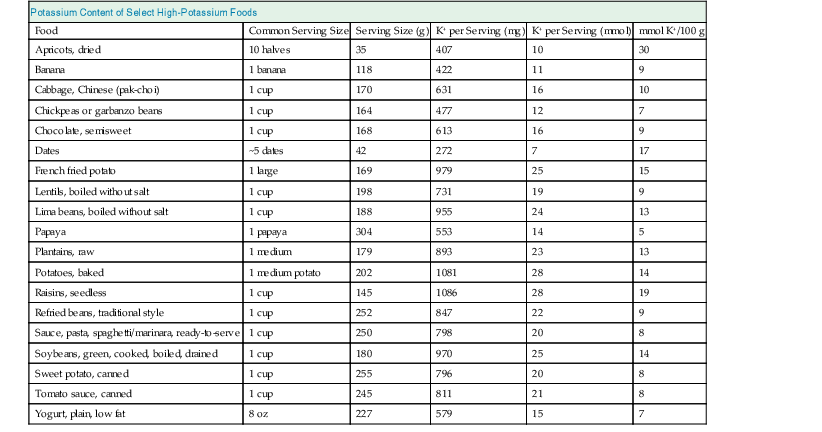
(Data from reference 1.)
Potassium Distribution
After absorption from the GI tract, potassium distributes into the extracellular fluid (ECF) and intracellular fluid (ICF) compartments. Potassium is the major intracellular cation, with K+ concentrations from about 100 to 120 mmol/l in the cytosol. Total intracellular K+ content is 3000 to 3500 mmol in healthy adults, found primarily in muscle (70%), with a lesser amount in bone, red blood cells, liver, and skin (Table 9-2). Only 1% to 2% total body potassium is present in the ECF. The electrogenic sodium pump, Na+-K+-ATPase, which is present in virtually all cells, affects this asymmetric potassium distribution by active cellular K+ uptake. Na+-K+-ATPase transports two potassium ions into cells in exchange for extrusion of three sodium ions, which results in high intracellular potassium concentration (high [K+]) and low intracellular sodium concentration (low [Na+]). Potassium-selective ion channels are the predominant determinant of the resting membrane potential. Therefore, the intracellular/extracellular [K+] ratio largely determines the resting cell membrane potential and the intracellular electronegativity. Normal maintenance of this ratio and membrane potential is critical for normal nerve conduction and muscular contraction.

Serum potassium is tightly regulated through multiple mechanisms. Recent studies support a “feed forward” regulatory system involving gut or portal potassium sensors. This system adjusts renal potassium excretion through mechanisms independent of serum potassium and aldosterone.2,3 This reflex system, which is still not understood fully, allows the kidney to “sense” dietary intake and alter renal potassium excretion despite no discernible changes in serum potassium or aldosterone concentration.
In addition, several hormones and factors can induce potassium shifts between the extracellular and intracellular potassium pools (Fig. 9-1). The most common causes include acid-base disorders, specific hormones, plasma osmolality, and exercise.
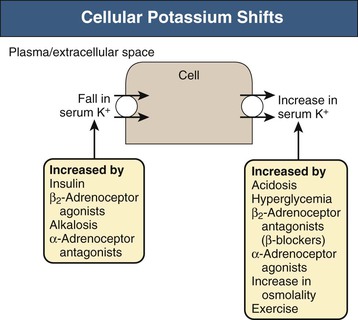
Acidosis caused by inorganic anions (e.g., NH4Cl, HCl) can cause hyperkalemia, but the mechanism is not fully understood. In contrast, organic acids (e.g., lactic acid) generally do not cause transcellular potassium shifts. Insulin and β2-adrenergic receptor activation induce cellular potassium uptake by stimulating Na+-K+-ATPase. Stimulation of Na+-K+-ATPase by insulin occurs through a mechanism separate from its stimulation of glucose entry. β2-Adrenoceptor activation increases intracellular cAMP production, which stimulates Na+-K+-ATPase-mediated potassium uptake, whereas α-adrenergic receptor activation has the opposite effect. The effects of insulin and β2-adrenoceptor activation are synergistic, as expected given the differing cellular mechanisms.
Aldosterone lowers serum potassium by two major mechanisms. First, aldosterone stimulates potassium movement into cells through stimulation of Na+-K+-ATPase (i.e., redistribution). Second, it can increase potassium excretion by the kidney and, to a lesser extent, by the GI tract. In the kidney, aldosterone stimulates sodium reabsorption in both the distal convoluted tubule and the collecting duct, and if there is sufficient sodium delivery to the collecting duct, this increased sodium reabsorption promotes potassium secretion. Mineralocorticoids can also stimulate potassium absorptive pumps, which may explain the equivocal effects of mineralocorticoids on potassium excretion despite their consistent effect on sodium absorption.4,5
Another important factor that alters cellular potassium uptake is plasma osmolality. Increased plasma osmolality, or hyperosmolality, when resulting from “effective osmoles,” can cause hyperkalemia. The likely mechanism is that increased plasma osmolality induces water movement out of the cells, which decreases cell volume and increases intracellular [K+]. This in turn results in feedback inhibition of Na+-K+-ATPase, decreasing cellular potassium uptake and normalizing intracellular [K+]. The clinician should remember that this occurs only with effective osmoles, such as with mannitol, or with hyperglycemia in diabetic patients. Both glucose in patients with intact insulin secretion and urea are “ineffective osmoles” because they rapidly cross plasma membranes and therefore do not alter cell volume. Importantly, administration of a glucose load to a nondiabetic patient, through stimulation of endogenous insulin secretion, can cause insulin-induced cellular potassium uptake and result in hypokalemia.
Exercise may result in hyperkalemia due to α-adrenergic receptor activation that shifts potassium out of the skeletal muscle cells. The increased serum potassium induces arterial dilation, which increases skeletal muscle blood flow and acts as an adaptive mechanism during exercise. Simultaneous β2-adrenoceptor activation stimulates skeletal muscle cellular potassium uptake and minimizes the severity of exercise-induced hyperkalemia, but this can lead to hypokalemia after cessation of exercise. In patients with preexisting potassium depletion, postexercise hypokalemia may be severe and can cause rhabdomyolysis.6
Renal Potassium Handling with Normal Renal Function
Long-term potassium homeostasis is accomplished primarily through changes in renal potassium excretion, almost entirely through regulated collecting duct potassium transport. Serum potassium is almost completely ionized, is not bound to plasma proteins, and is filtered efficiently by the glomerulus (Fig. 9-2). The proximal tubule reabsorbs the majority (~65% to 70%) of filtered potassium, but there is relatively little variation in proximal tubule potassium reabsorption in response to hypokalemia or hyperkalemia. In the loop of Henle, potassium is secreted in the descending loop, at least in deep nephrons, and is reabsorbed in the ascending loop through the action of the Na+-K+-2Cl− cotransporter (Fig. 9-3, A). However, the majority of K+ transported by this protein is recycled back into the tubular lumen through an apical K+ channel. As a result, there is only modest net potassium reabsorption in the Henle loop. This absorption can be reversed to secretion, however, by administration of a loop diuretic or by substantial potassium loading. However, the magnitude of the change in Henle loop potassium transport in various physiologic conditions is rather small. Consequently, potassium excretion is regulated under most conditions through variations in distal convoluted tubule and collecting duct potassium transport.
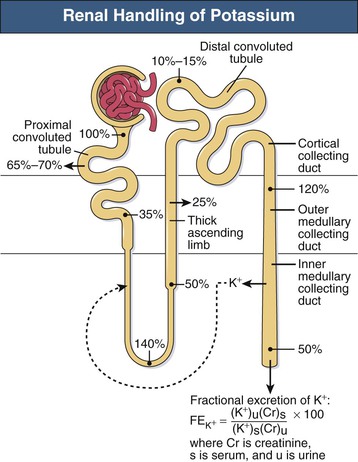
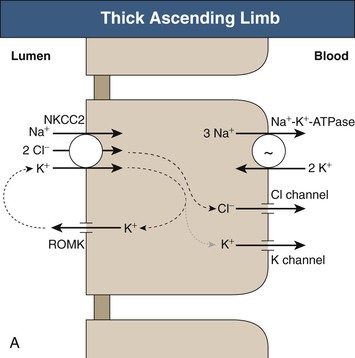
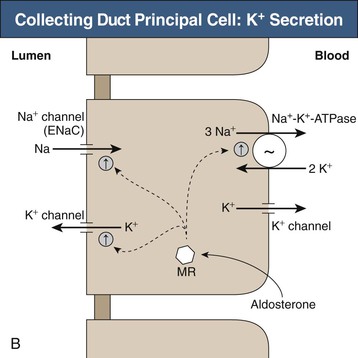
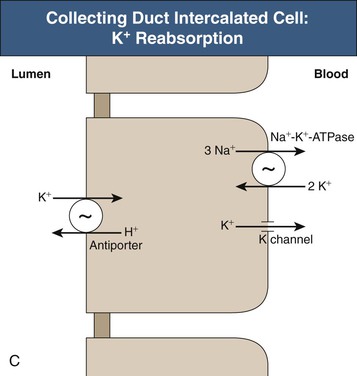
The collecting duct consists of two fundamentally different cell types with distinctly different roles in potassium transport. The principal cell in the cortical collecting duct secretes potassium, whereas intercalated (IC) cells appear to reabsorb potassium. In the principal cell, sodium is reabsorbed through the apical epithelial sodium channel (ENaC), which stimulates basolateral Na+-K+-ATPase (Fig. 9-3, B); active potassium uptake by this protein maintains a high intracellular [K+]. Subsequent to basolateral potassium uptake, K+ is secreted across the apical plasma membrane of principal cells into the luminal fluid by apical potassium channels and KCl cotransporters. IC cells, in contrast, reabsorb potassium through an apical H+-K+-ATPase7 (Fig. 9-3, C); this protein actively secretes H+ into the luminal fluid in exchange for reabsorption of luminal potassium. The presence of two separate potassium transport processes, secretion by principal cells and reabsorption by IC cells, enables rapid and effective regulation of renal potassium excretion.
Several factors regulate principal cell potassium secretion and, in relative order of importance, include luminal flow rate, distal sodium delivery, aldosterone, extracellular potassium, and extracellular pH. An increase in luminal flow rate reduces luminal [K+], thereby increasing the concentration gradient across the apical membrane, which stimulates potassium secretion. In addition, flow rate directly influences cellular potassium secretion, possibly by modulating the activity of potassium channels. Conversely, reduced luminal flow, such as occurs in pre-renal azotemia and obstruction, may result in hyperkalemia. Decreased sodium reabsorption, whether from reduced luminal sodium delivery or sodium channel inhibitors, decreases K+ secretion by altering electrochemical forces for K+ secretion. “Potassium-sparing diuretics” function by blocking principal cell ENaC-mediated sodium reabsorption, which inhibits principal cell potassium secretion. Conversely, increased sodium delivery to the collecting duct, as may occur with loop or thiazide diuretics, increases principal cell sodium reabsorption and causes a secondary increase in potassium secretion. Aldosterone has many effects that increase principal cell potassium secretion, including increases in Na+-K+-ATPase expression and increased apical expression of eNaC. The net effect is increased K+ secretion. Increasing extracellular potassium directly stimulates Na+-K+-ATPase activity, leading to increased K+ secretion. Metabolic acidosis decreases K+ secretion, both through direct effects on potassium channels and through changes in interstitial ammonia concentration, which decreases K+ secretion.8 Respiratory acidosis has minimal effect on K+ secretion, whereas chronic respiratory alkalosis causes marked increases in K+ excretion because of increases in urinary bicarbonate excretion.
Potassium reabsorption, which occurs in parallel with potassium secretion, decreases net renal K+ excretion, and occurs through the action of the potassium-reabsorbing protein, H+-K+-ATPase. The major factors regulating H+-K+-ATPase expression and activity include potassium balance, aldosterone, and acid-base status. Potassium depletion increases H+-K+-ATPase expression, resulting in increased active potassium reabsorption and decreased potassium excretion. Aldosterone also increases H+-K+-ATPase expression and activity and, by decreasing net potassium excretion, may function to minimize the hypokalemia that otherwise results from aldosterone’s effect on both potassium redistribution and principal cell–mediated K+ secretion. Metabolic acidosis has both direct and indirect effects, mediated through alterations in ammonia metabolism, that increase H+-K+-ATPase potassium transport. In some patients, this may contribute to the hyperkalemia that can occur with metabolic acidosis.
An important class of intracellular signaling proteins have been identified that are fundamental in the regulation of renal K+ transport in the distal nephron. The “with no lysine” or “WNK” kinases are a family of proteins expressed in many cells of the body including the kidney. Under basal conditions, WNK kinases inhibit Na+ reabsorption as well as decreasing potassium secretion, in part by inhibiting the rectifying renal outer medulla potassium (ROMK) channel. Genetic defects that inactivate WNK kinases result in enhanced Na+ reabsorption and reduced K+ secretion.
Renal Potassium Handling in Chronic Kidney Disease
Potassium homeostasis is relatively well preserved and serum potassium usually remains in the normal range until glomerular filtration rate is reduced substantially. This adaptation is due largely to increased rates of per nephron potassium excretion in the connecting segment and the collecting duct. Both aldosterone and an increase in serum potassium may contribute to this adaptation. Intestinal potassium secretion increases, also, although quantitatively this is less important.
Patients with chronic kidney disease (CKD) have more difficulty handling an acute potassium load, even when they have a normal serum [K+]. Because these patients have decreased nephron number, their maximal capacity for K+ secretion is limited. Patients with CKD are also routinely treated with medications that alter renal potassium handling, such as angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and β-blockers. These can decrease renal potassium sensitivity and result in higher serum potassium concentrations.
Patients with CKD generally tolerate hyperkalemia with fewer cardiac and electrocardiographic (ECG) abnormalities than do patients with normal renal function. The mechanism of this adaptation is incompletely understood. In particular, patients with CKD appear to tolerate serum [K+] of 5.0 to 5.5 mmol/l with no significant adverse effect, and levels of 5.5 to 6.0 mmol/l are associated with lower mortality than [K+] of 3.5 to 3.9 mmol/l.9 Nevertheless, severe hyperkalemia (>6.0 mmol/l or presence of ECG changes) can have lethal effects and should be treated aggressively.
Hypokalemia
Epidemiology
The incidence of potassium disorders depends greatly on the patient population. Less than 1% of adults with normal renal function who are not receiving medicines develop hypokalemia or hyperkalemia; however, diets with high sodium and low potassium content may lead to potassium depletion. Thus, identification of hypokalemia or hyperkalemia should suggest either that an underlying disease is present or that the individual is taking medications that alter potassium handling. For example, hypokalemia may be present in as many as half of patients taking diuretics10 and is present frequently in patients with primary or secondary hyperaldosteronism.
Clinical Manifestations
Potassium deficiency, because it alters the ratio of extracellular to intracellular potassium, alters the resting membrane potential, which can impair normal functioning of almost every cell in the body. This particularly is true for cells in the heart and blood vessels, nerves, muscles, gut, and kidneys. Overall, children and young adults tolerate hypokalemia better than elderly persons. Prompt correction is warranted in patients with coronary heart disease or in patients receiving digitalis glycosides, because of an increased risk of lethal cardiac arrhythmias.
Cardiovascular
Epidemiologic studies link hypokalemia and a low-potassium diet with an increased prevalence of hypertension. Experimental studies show that hypokalemia increases blood pressure by 5 to 10 mm Hg, and that potassium supplementation can lower blood pressure by a similar amount.11 Potassium deficiency increases blood pressure through multiple mechanisms, including stimulating sodium retention and causing intravascular volume expansion, and by sensitizing the vasculature to endogenous vasoconstrictors.11 In part, sodium retention is related to decreased expression of the kidney-specific isoform of WNK1, which leads to increased NaCl cotransporter (NCC)–mediated and ENaC-mediated sodium reabsorption in the distal convoluted tubule and cortical collecting duct, respectively.12
Hypokalemia increases the risk of a variety of ventricular arrhythmias, including ventricular tachycardia and ventricular fibrillation.13 Diuretic-induced hypokalemia is of particular concern, because sudden cardiac death may occur more frequently in those treated with thiazide diuretics.13 Ventricular arrhythmias are also more common in patients receiving digoxin.
Hormonal
Hypokalemia both impairs insulin release and induces insulin resistance, resulting in worsened glucose control in diabetic patients.14 The insulin resistance that usually occurs with thiazide diuretic therapy is caused by endothelial dysfunction mediated by thiazide-induced hypokalemia and hyperuricemia.15,16
Muscular
Hypokalemia hyperpolarizes skeletal muscle cells, thereby impairing muscle contraction. Hypokalemia also reduces skeletal muscle blood flow, possibly by impairing local nitric oxide release; this effect can predispose patients to rhabdomyolysis during vigorous exercise.17
Renal
Hypokalemia leads to several important disturbances of renal function. Reduced medullary blood flow and increased renal vascular resistance may predispose to hypertension, tubulointerstitial and cystic changes, alterations in acid-base balance, and impairment of renal concentrating mechanisms.
Potassium depletion causes tubulointerstitial fibrosis that is generally greatest in the outer medulla. The degree of reversibility is related to the duration of hypokalemia, and if prolonged, hypokalemia may result in renal failure. Experimental studies suggest increased risk for irreversible renal injury when hypokalemia is present during the neonatal period.18 Longstanding potassium depletion also causes renal hypertrophy and predisposes to renal cyst formation, particularly when there is increased mineralocorticoid activity.
Metabolic alkalosis is a common acid-base consequence of potassium depletion and results from increased renal net acid excretion caused by increased renal ammonia excretion.19 Conversely, metabolic alkalosis may increase renal potassium excretion and cause potassium depletion. Severe hypokalemia can lead to respiratory muscle weakness and development of respiratory acidosis.
Severe hypokalemia also impairs concentrating ability, causing mild polyuria, typically 2 to 3 l/day. Both increased thirst and mild nephrogenic diabetes insipidus contribute to the polyuria.20 The nephrogenic diabetes insipidus is caused by decreased expression of several proteins, such as the water transporter aquaporin 2 (AQP2), and the urea transporters UT-A1, UT-A3, and UT-B, which are involved in urine concentration and water reabsorption.
Hypokalemia has substantial effects to increase renal ammonia production. Some ammonia is excreted in the urine, increasing net acid excretion and leading to development of metabolic alkalosis. In addition, approximately half of this increase returns to the systemic circulation via the renal veins. In patients with acute or chronic liver disease, this increased ammonia delivery may exceed hepatic ammonia clearance capacity, increase plasma ammonia levels, and either precipitate or worsen hepatic encephalopathy.21
Etiology
Hypokalemia results typically from one of four etiologies: pseudohypokalemia, redistribution, extrarenal potassium loss, or renal potassium loss. However, multiple etiologies may coexist in the specific patient.
Pseudohypokalemia
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


