PATHOGENESIS OF UREMIC BLEEDING
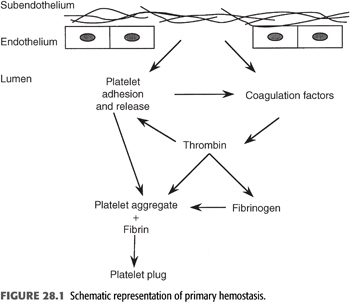
The causes of uremic bleeding have been the subject of a major debate since the 1970s. Most landmark studies in the field were indeed conducted in the 1970s to 1980s, when, in the early days of dialysis, hemorrhagic issues often posed major clinical challenges. In the following decades, widespread adoption of erythropoiesis-stimulating agents substantially reduced the clinical relevance of uremic bleeding. Nonetheless, a deep understanding of the hemostatic derangements in ESKD remains pivotal to provide optimal patient care. The pathogenesis of uremic bleeding is multifactorial, and the major defects involve primary hemostasis (FIGURE 28.1), that is, platelet–vessel wall and platelet–platelet interactions.
Platelet Abnormalities and Uremic Toxins
Moderate thrombocytopenia is commonly found in uremic patients, suggesting inadequate production or platelet overconsumption (6). Nonetheless, thrombocytopenia rarely is severe enough to cause bleeding. Numerous biochemical changes have been reported in platelets from ESKD patients. Dense granule content is altered in uremic platelets, with reduced levels of the aggregation agonists serotonin and adenosine diphosphate (ADP) (7). Granule secretion is defective too: Release of adenosine triphosphate (ATP) from dense granules (8) and of α-granule proteins, including β-thromboglobulin (9), are impaired. Calcium content is increased in platelets from uremic patients (10), and its mobilization in response to stimulation is abnormal (11), leading to functional defects. Anomalies in the platelet cytoskeleton contribute to defective hemostasis in chronic kidney disease (CKD): Cytoskeletal proteins are quantitatively reduced, and their assembly upon stimulation is qualitatively abnormal (12), hindering cellular contraction and motility. Platelets from uremic patients also display defects in two adhesion receptors, glycoprotein (Gp) Ib and the Gp IIb–IIIa complex. Gp Ib is fundamental for platelet adhesion to the subendothelium at high shear rates, like those found in the capillary circulation. Availability of Gp Ib on the platelet surface is decreased in CKD, likely because of enhanced proteolysis of the receptor (13). Platelet adhesion to subendothelial surfaces leads to activation of the Gp IIb–IIIa receptor complex, that in turn binds von Willebrand factor (vWF) and fibrinogen. The number of Gp IIb–IIIa receptors expressed on the platelet membrane of uremic patients is normal, but their function is impaired (14,15). This has been ascribed to competitive binding of the receptor by fibrinogen fragments and, possibly, other dialyzable toxic substances that accumulate in uremic plasma.
Arachidonic acid and prostaglandin metabolism is deregulated in uremia: A relative deficiency of the platelet agonist thromboxane A2 and an excess of the potent platelet inhibitor and vasodilator prostacyclin are found, tipping the balance toward defective platelet function. Production of thromboxane A2 by platelets in response to endogenous and exogenous stimuli is impaired (16,17). On the other hand, formation of prostacyclin is increased in uremic patients (18), and their plasma contains higher than normal amounts of a factor that stimulates vascular production of prostacyclin (19). Elevation in prostacyclin and pathologic hyperactivity of platelet adenylate cyclase (20) cooperate in increasing intracellular levels of cyclic adenosine monophosphate (cAMP) (21), which in turn contributes to impaired platelet function.
Nitric oxide (NO) is another key player in uremic bleeding (22,23) that interferes with hemostasis by inhibiting platelet aggregation and by preventing the vasoconstriction that normally follows vessel injury. Rats made uremic by extensive surgical ablation of kidney mass display bleeding time prolongation and elevated plasma levels of the stable NO metabolites nitrites and nitrates (24). In this animal model, increased expression of both inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) is found in the aorta (24), consistently with enhanced vascular release of NO. Higher plasma levels of L-arginine, the substrate for NO synthesis, have been documented in uremic patients, compared with healthy volunteers. Platelets taken from CKD patients generate more NO than control platelets (25), and uremic plasma, unlike plasma from controls, potently induces in vitro NO production in cultured human umbilical vein or microvascular endothelial cells (26). Increased NO synthesis is likely the main mechanism by which the uremic toxins guanidinosuccinic acid and methylguanidine interfere with platelet function (26). Cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), which rise in ESKD, are also potent inducers of iNOS (27).
After mixing with plasma from healthy subjects, uremic platelets show improvement of many functional parameters, while mixing of uremic plasma with normal platelets leads to platelet dysfunction. These findings highlight the key role of soluble plasma factors in determining platelet defects. The already cited elevation in prostacyclin, fibrinogen fragments, and inducers of NO synthesis, together with reduced thromboxane A2 in uremia, partially account for these effects. The metabolites phenol, phenolic acid (28), and guanidinosuccinic acid (29) rise in CKD and impair platelet aggregation to ADP in vitro. Also, parathyroid hormone (PTH) has been consistently shown to inhibit platelets in vitro (30). Nonetheless, a clear correlation between skin bleeding time and the serum concentrations of these substances has not been demonstrated in ESKD patients (31,32). Other uremic retention solutes, like urea, creatinine, and guanidinoacetic acid, do not significantly interfere with platelet function when added to normal plasma (33).
Chronic volume overload in dialysis patients has been proposed to be an additional factor that increases bleeding risk, in particular of spontaneous subdural hematomas. The putative pathogenic mechanism entails venous hypertension, predisposing to small venous tears of dural bridging veins (4).
Anemia
Anemia is a common finding in ESKD patients and has a multifactorial origin, encompassing shortened survival of red blood cells in the uremic milieu, reduced responsiveness of the erythroid marrow induced by uremic toxins (34), repeated blood loss during dialysis, and, most importantly, defective secretion of erythropoietin. Anemia plays a key role in uremic bleeding, mainly through rheologic factors. At a low hematocrit, platelets tend to be dispersed along the blood vessels, which impairs their interaction with the vascular walls (35). At a hematocrit above 30% instead, erythrocytes mainly localize at the center of the vessels and platelets are displaced more peripherally, closer to the endothelial surface where their efficiency in forming a plug, in case of exposure of the subendothelium, is optimized. Erythrocytes also directly enhance platelet function by releasing platelet aggregation agonists ADP and thromboxane A2, by inactivating prostacyclin, and by scavenging NO through hemoglobin (35).
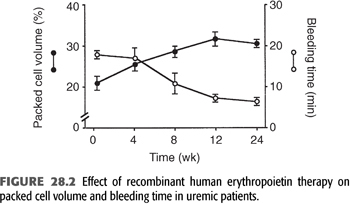
Skin bleeding time is inversely correlated with hematocrit and significantly shortened after red blood cell transfusion (36,37). Treatment with recombinant human erythropoietin also reduces bleeding time and increases hemostatic efficiency (38). In a randomized study, erythropoietin normalized the bleeding time in all uremic patients as hematocrits rose from 27% to 32% (39).
Effects of Dialysis
Dialysis has opposing effects on hemostasis. On the one hand, it allows removal of some of the uremic toxins that induce platelet dysfunction and hemostatic derangements. As a proof of concept, for example, skin bleeding time is significantly shortened, albeit rarely normalized, in uremic patients after the first dialysis sessions (40). However, dialysis cannot effectively remove all uremic retention solutes, especially those with high molecular weight or protein-bound, and only partially corrects uremia and the associated bleeding tendency (31).
Importantly, dialysis itself can increase bleeding risk, especially in the case of hemodialysis, where blood gets in contact with the extracorporeal circuit and systemic anticoagulation is usually required (41). In patients on chronic hemodialysis, skin bleeding time is often longer right after the dialysis session than before, an effect not attributable to heparin use (42). In spite of significant advances in biocompatibility with current hemodialysis technology, exposure of blood to the dialyzer membrane, to circuit tubing, to microbubbles in the circuit, and to shear stress, particularly in the roller pump segment, inevitably leads to a certain degree of activation of platelets, inflammatory cells, coagulation, and complement system. As well documented also in the setting of cardiopulmonary bypass, continual platelet activation can bring about mild thrombocytopenia and transient functional platelet exhaustion due to reiterated degranulation and loss of Gp receptors (41).
In the course of the hemodialysis session, hemostasis is modulated also by oscillations in the plasma levels of NO, which are the result of two opposing processes. Hemodialysis removes uremic toxins that stimulate NO synthesis and, at the same time, activates inflammation pathways in the extracorporeal circuit, raising the levels of potent NO inducers like the cytokines TNF-α and IL-lβ (25,43). In particular, NO induction has been attributed to complement-activating dialyzer membranes, acetate-containing dialysate, and dialysate contamination by intact endotoxin, endotoxin fragments, and other bacterial components that may cross the dialysis membranes. The current use of synthetic, more biocompatible, membranes, the widespread availability of bicarbonate dialysis, and the adoption of more and more stringent standards of dialysate purity could hopefully tip the balance toward a net reduction in NO during hemodialysis, thus improving hemostasis.
Administration of anticoagulants to avoid circuit clotting during hemodialysis is an additional factor that can increase bleeding risk. In particular, heparin, the drug most widely used for this purpose, is known to promote platelet self-aggregation and activation through nonimmunogenic mechanisms, which can exacerbate the functional alterations generated by the extracorporeal circulation.
From the standpoint of bleeding risk, peritoneal dialysis presents clear theoretical advantages over hemodialysis by avoiding the need of intradialytic anticoagulation and the exposure of blood to the extracorporeal circuit. Along the same line, epidemiologic studies have documented a lower incidence of gastrointestinal and intracranial bleeding in peritoneal dialysis, compared to hemodialysis patients (5,44).
Medications
Dialysis patients are often prescribed anticoagulant and antiplatelet agents due to their cardiovascular conditions or to preserve vascular access. Compared to the general population, these drugs put uremic patients at higher bleeding risk (45,46), exacerbating their hemorrhagic diathesis. Particular caution must be exercised prescribing drugs like low molecular weight heparins, which rely on a primarily kidney metabolism and tend to accumulate in ESKD (47). Bleeding risk due to bioaccumulation is significantly raised also with the new oral anticoagulants, targeting either thrombin (e.g., dabigatran) or factor Xa (e.g., apixaban), which are contraindicated in dialysis (48). The effects of aspirin on hemostasis are potentiated in the uremic milieu: In addition to the well-known irreversible blocking of platelet cyclooxygenase, aspirin also causes a cyclooxygenase-independent marked prolongation of the bleeding time in uremic patients but not in normal subjects (49). In addition, aspirin and the other nonsteroidal anti-inflammatory drugs directly hamper integrity of the gastrointestinal mucosa through inhibition of prostaglandin synthesis (50). This effect may be of particular relevance in dialysis patients, that are especially prone to gastrointestinal bleeding.
Other drugs that can contribute to uremic bleeding are β-lactam antibiotics. They accumulate in advanced kidney disease and may perturb platelet membrane, interfering with the ADP receptor and therefore inhibiting platelet aggregation (51). These effects are dose- and duration-dependent and resolve after drug discontinuation. Among the β-lactam antibiotics, third-generation cephalosporins may affect hemostasis the most, due to concomitant interference with intrahepatic vitamin K metabolism, in addition to platelet inhibition (52).
 CLINICAL FINDINGS
CLINICAL FINDINGS
The most common bleeding complications in uremia are petechial hemorrhages, blood blisters, and ecchymoses at the site of fistula puncture or temporary venous access insertion. Less frequent but more serious bleeding problems are discussed below.
Gastrointestinal Bleeding
The gastrointestinal tract is the most frequent site of major bleeding in ESKD patients (53). The sources of gastrointestinal hemorrhage are similar to those in patients with normal kidney function and include gastric and duodenal ulcers/erosions, vascular ectasias, esophagitis, gastritis, Mallory-Weiss tears, gastric cancer (54), hemorrhoids, colonic diverticular disease, and polyps (55).
Angiodysplasias (telangiectasias, arteriovenous malformations) are small vascular lesions of the gastrointestinal mucosa and submucosa that have been observed all along the digestive tract and may cause acute or subacute hemorrhage. They have been reported as a cause of bleeding more frequently in dialysis patients than in the general population (56). It is not clear whether this finding reflects a higher incidence of angiodysplasias in the setting of CKD and/or an increased risk for these lesions to bleed and thus to be diagnosed in uremic patients.
Ischemic colitis is another important differential diagnosis in dialysis patients with lower gastrointestinal bleeding, especially in those with widespread vascular disease and prone to intradialytic hypotension (57).
Alternative rare causes of digestive hemorrhage, like Kaposi’s sarcoma, cytomegalovirus colitis, and non–Hodgkin’s lymphoma, need to be considered in patients with concomitant severe immunodepression, such as those with HIV nephropathy or a history of long-term exposure to immunosuppressant drugs for kidney transplantation or immune-mediated systemic diseases (55).
Intracranial Hemorrhage
ESKD patients are more prone than the general population to intracranial bleedings, including subdural hematomas, intracerebral hemorrhages, and subarachnoid hemorrhages (4,58,59). Such events are often spontaneous, without recallable trauma, and are associated with higher mortality in dialysis patients. For instance, data from a large U.S. database showed that the incidence rate of subarachnoid hemorrhages was more than sixfold higher in patients on dialysis, who had a mortality of 38.4% versus 21.9% in the general population (59).
Brain magnetic resonance imaging (MRI) studies have also shown that small hemosiderin deposits, indicative of prior microscopic cerebral hemorrhage, are highly prevalent among hemodialysis patients. These alterations, known as cerebral microbleeds, are closely associated with other cerebral small-vessel diseases and likely contribute to cognitive dysfunction; age and high blood pressure are significant risk factors (60,61).
Hemorrhagic Pericarditis and Pleuritis
Limited, fibrinous pericardial and pleural effusions are common findings in patients with advanced kidney disease. Without adequate dialysis, serous effusion can undergo hemorrhagic transformation, leading to life-threatening uremic pericarditis, with possible tamponade (62), or pleuritis (63). These presentations used to be frequent in the predialysis era and are now rarely encountered. Nonetheless, they remain medical emergencies that require prompt recognition and treatment, the core of management being intensive, anticoagulant-free dialysis.
Other Hemorrhagic Presentations
Women on dialysis frequently present abnormal uterine bleedings, with a high prevalence of uncontrolled menorrhagia, contributing to symptomatic anemia (64). Kidney cysts are a common finding in dialysis-associated cystic kidney disease and occasionally give rise to unprovoked bleeding. Presentations range from asymptomatic, trivial intracystic hemorrhage to massive retroperitoneal hematomas (65).
Subcapsular liver hematoma is another serious hemorrhagic complication that has been rarely observed in uremic patients (66).
Notably, patients with CKD from autosomal dominant polycystic kidney disease may be at higher than average bleeding risk, due to predisposing anatomic abnormalities like kidney and liver cysts, an elevated frequency of colon diverticulosis and susceptibility to develop intracranial aneurysms (67).
Finally, in spite of the current improvements in standards of care, major surgery and invasive procedures still entail perioperative hemorrhagic risks that are more elevated in dialysis patients than in the general population, with subsequent higher morbidity and mortality rates (68).
 LABORATORY ASSESSMENT OF THE BLEEDING RISK
LABORATORY ASSESSMENT OF THE BLEEDING RISK
The single assay that best correlates with uremic bleeding is skin bleeding time, an in vivo test that gives an overall assessment of primary hemostasis (69). Among the factors that influence skin bleeding time, there are platelet number and functionality, hematocrit, vascular integrity, and activity of vWF. The skin bleeding time is most commonly determined using the Ivy method. A blood pressure cuff is applied above the cubital fossa and inflated at 40 mm Hg to elevate venous pressure and increase test sensitivity. A small cut is made on the ventral side of the subject’s forearm, in an area where there are no hairs or visible veins. The cut is done quickly by an automatic device, which provides a standardized width and depth. The blood is blot away every 30 seconds by filter paper, until the platelet plug forms and the bleeding terminates. The time it takes for the bleeding to stop is measured and represents the skin bleeding time. Normal values are below 7 minutes; prolongation above 10 minutes correlates with a significantly increased risk of bleeding in uremic patients. The skin bleeding time was established as a predictor of hemorrhagic events in CKD patients in the 1970s and suffers from poor reproducibility and accuracy; nonetheless, it remains the best available test at present.
Several in vitro assays of platelet function have been developed, such as closure time test (also known as platelet function analyzer or PFA-100, that measures platelet aggregation in response to ADP and epinephrine), whole blood platelet aggregation (WBPA), thromboelastography (TEG), and cone platelet analyzer (CPA) (70). However, none of these tests has been validated in clinical practice in the setting of uremic bleeding, and their use remains investigational.
 PREVENTION AND TREATMENT
PREVENTION AND TREATMENT
We will focus on the following key points that should be systematically addressed in all dialysis patients in order to prevent bleeding and optimize treatment of active hemorrhage (TABLE 28.1): the dialysis prescription, correction of anemia, appropriateness of concomitant antiplatelet/anticoagulant therapies, and use of specific agents to improve hemostasis.

In case of ongoing hemorrhage, first-line management is not specific for dialysis patients and includes hemodynamic support; identification of the hemorrhagic source and, possibly, subsequent treatment by surgical, endovascular, or endoscopic techniques. ESKD should be accounted for in the risk assessment of bleeding patients and in the subsequent planning of therapeutic interventions. For example, markedly higher rebleeding rates from endoscopically treated peptic ulcers have been documented in dialysis patients, even when these patients had low-risk ulcer stigmata on endoscopy (71). While awaiting more comprehensive data, it could thus be reasonable to manage all dialysis patients with peptic ulcer bleeding, and maybe other hemorrhagic disorders as well, as high-risk patients.
Dialysis Prescription
Adequate dialysis is a mainstay in the prevention of uremic bleeding. However, as already discussed, current dialysis technologies allow only partial correction of the bleeding diathesis associated with uremia, and hemodialysis in particular interferes with hemostasis. For these reasons, in the absence of other contraindications, peritoneal dialysis is a reasonable choice for patients at particularly high hemorrhagic risk.
In the hemodialysis prescription, intradialytic anticoagulation needs to be personalized, balancing individual thrombotic and hemorrhagic risks. The minimum dose of heparin effective in preventing circuit clotting should be determined for each patient. Reduced-intensity intradialytic anticoagulation is a sensible option in patients treated with oral anticoagulants: In many of such cases, even complete avoidance of intradialytic anticoagulation is feasible, provided that the international normalized ratio (INR) is within the target range, there is minimal inflammation, a native vascular access with adequate blood-flow rate is available, and synthetic high-flux membranes are used (72). As detailed elsewhere in this book, use of minimum-dose heparin or no-heparin hemodialysis is possible in patients at high bleeding risk or with ongoing hemorrhage, at the cost of increasing the possibilities of circuit clotting. Regional citrate anticoagulation is a valid alternative. In case of life-threatening bleeding, like intracranial hemorrhages, complete avoidance of anticoagulants is advised, usually for a period of at least 2 weeks.
Dialytic adequacy must be carefully considered, especially in patients presenting with uremic or dialysis-associated pericarditis. In this setting, it is mandatory to make a trial of intensive dialysis, usually defined, in the case of hemodialysis, as daily sessions for 10 to 15 days, preferably without heparin to minimize the risk of hemopericardium and cardiac tamponade (62).
In case central venous catheters are used as hemodialysis access, heparin lock is an often under recognized factor possibly concurring to bleeding risk. In fact, a certain degree of spillage of lock solution in the systemic circulation is practically inevitable, and if high-concentration heparin (5,000 to 10,000 U/mL) is employed, this can occasionally contribute to clinical bleeding. Use of 4% citrate or low-dose heparin (1,000 U/mL) is safe and effective alternative as lock solution (73,74).
Correction of Anemia
Management of anemia is pivotal in the prevention and treatment of uremic bleeding. Use of recombinant human erythropoietin has largely replaced transfusions and significantly lowered hemorrhagic risk. A randomized study established that a threshold hematocrit between 27% and 32% effectively normalized bleeding time in uremic patients on erythropoietin (FIGURE 28.2) (39). Treatment of anemia likely improves hemostasis by displacing platelets from a midstream position toward the vessel wall, thus encouraging platelet–endothelium interactions. In addition, recombinant human erythropoietin affects hemostasis also independently of amelioration of anemia. For instance, it increases the number of reticulated platelets, which are more metabolically active (75), improves platelet calcium signaling (76), and enhances thrombin-induced phosphorylation of platelet proteins (77).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


