Neonatal hepatitis
Cholestatic syndromes
Peroxisomal disorders
Idiopathic NH
PFIC
Zellweger
Viral NH
Type 1 Byler P-type ATPase
Infantile Refsum
CMV
Type 2 canalicular Bile Acid Tx
Other enzymopathies
Herpes
Type 3 MDR3 deficiency
Bile acid synthetic disorders
Rubella
Aagenaes cholestasis lymphedema
3b-hydroxysteroid dehydrogenase
Reovirus
N. Am. Indian cholestasis
D4-3-oxosteroid 5b-reductase
Adenovirus
Nielsen Greenland Eskimo cholestasis
Oxosterol 7a-hydroxylase
Enteroviruses
Benign recurrent intrahepatic cholestasis
Toxic
Parvovirus B19
Dubin-Johnson MRP2 cMOAT deficiency
Drugs
Paramyxovirus
Rotor syndrome
Parenteral alimentation
Hepatitis B
Metabolic disorders
Aluminum
HIV
a1-Antitrypsin deficiency
Miscellaneous associations
Bacterial and parasitic
Cystic fibrosis
Shock/hypoperfusion
Bacterial sepsis
Neonatal iron storage disease
Histiocytosis X
UTI
Endocrinopathies
Neonatal lupus erythematosus
Syphilis
Hypopituitarism
Indian childhood cirrhosis
Listeriosis
Hypothyroidism
Autosomal trisomies 17, 18, 21
Toxoplasmosis
Amino acid disorders
Graft-versus-host disease
Tuberculosis
Tyrosinemia
Malaria
Hypermethionemia
Bile duct obstruction
Mevalonate kinase deficiency
Cholangiopathies
Lipid disorders
Biliary atresia
Niemann-Pick A, B
Choledochal cysts
Niemann-Pick C
Non-syndromic paucity
Gaucher
Alagille syndrome
Wolman
Sclerosing cholangitis
Cholesterol ester storage disease
Spontaneous duct perforation
Urea cycle disorders
Caroli’s disease
Arginase deficiency
Congenital hepatic fibrosis
Carbohydrate disorders
Bile duct stenosis
Galactosemia
Other
Fructosemia
Inspissated bile/mucus
Glycogen storage IV
Cholelithiasis
Mitochondrial disorders
Tumors
Oxidative phosphorylation
Masses
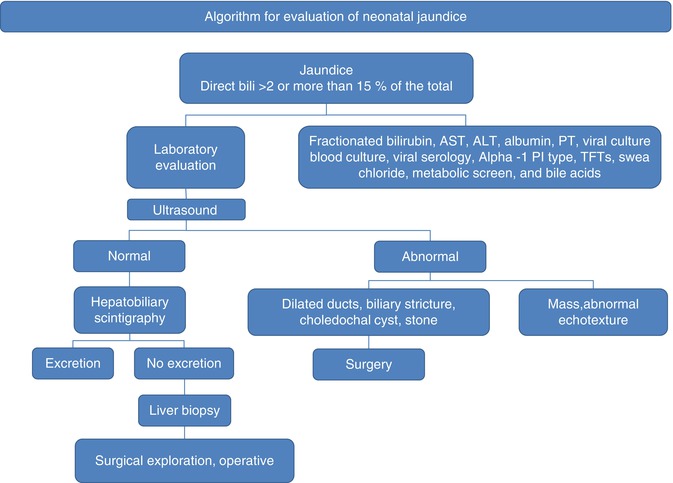
Fig. 10.1
Algorithm for the evaluation of the infant with jaundice
Table 10.2
Estimated frequency of various clinical forms of neonatal cholestasis
Clinical form | Percentage |
|---|---|
Idiopathic neonatal hepatitis | 30–35 |
Extrahepatic biliary atresia | 25–30 |
Alpha-1-antitrypsin deficiency | 7–10 |
Intrahepatic cholestasis syndrome (Alagille, Progressive familial cholestasis, etc.) | 5–6 |
Bacterial sepsis | |
Hepatitis | 2 |
Cytomegalovirus | 3–5 |
Rubella, herpes | 1 |
Endocrine (hypothyroidism, panhypopituitarism) | 1 |
Galactosemia | 1 |
Inborn errors of bile acid biosynthesis | 2–5 |
However, much of the sequence of tests is driven by consideration of biliary atresia (BA), as this disorder accounts for about 30 % of cases of neonatal cholestasis and it is known that children with BA have a better surgical outcome with early intervention. As laboratory findings in this young age group do not have much discriminatory value, ultrasound and hepatobiliary scintigraphy are obtained to try to identify correctable causes of jaundice. The ultrasound is an optimal tool for diagnosing choledochal cyst or stone, and scintigraphy can be useful to assess patency of the biliary tract (tracer reaches the intestine) or possible obstruction (no tracer reaches the intestine despite adequate update of tracer by hepatocytes). However, even these findings can be unreliable and the diagnostic evaluation proceeds to liver biopsy to determine if surgical exploration of the biliary tree is warranted.
10.4 Cholestasis in the Older Child
In the older child, the laboratory tests provide better discriminatory value between intrahepatic and extrahepatic processes. Elevations of conjugated bilirubin and/or bile acids are accompanied by relatively mild or even absent elevations of hepatic enzymes in diseases such as cholecystitis. Ultrasound is more likely to detect duct dilatation which is virtually never seen in the infant liver despite complete obstruction. The differential diagnosis is relatively small compared to that in the neonate; therefore, the evaluation is much more directed and the pathologist is less frequently involved. The major considerations include hepatitis (infectious, autoimmune, toxic), gallstones, hydrops, stricture, sclerosing cholangitis, choledochal cyst, and masses (Table 10.3 ).
Table 10.3
Differential diagnosis of cholestasis in the older child and young adult
Extrahepatic cholestasis | Cholelithiasis |
Tumors, e.g., cholangiocarcinoma | |
Primary sclerosing cholangitis (late presentation) | |
AIDS cholangiopathy | |
Pancreatitis – acute and chronic | |
Strictures | |
Parasites (Ascaris, liver flukes) | |
Intrahepatic cholestasis | AHD (late cholestatic presentation) |
Viral hepatitis | |
NASH | |
Drugs/toxins (alkylated steroids, chlorpromazine, herbal medications such as bush tea, arsenic) | |
Sepsis and hypoperfusion states | |
Infiltrative disease (amyloid, lymphoma, sarcoid, tuberculosis) | |
TPN | |
Hepatic crisis in sickle cell disease | |
Pregnancy | |
Wilson’s disease | |
Choledochal cyst | |
BRIC | |
Autoimmune hepatitis | |
Jaundice without cholestasis | Dubin-Johnson syndrome |
Rotor syndrome |
10.5 Biliary Atresia
10.5.1 General Aspects
Biliary atresia (BA) can be defined as a progressive fibroinflammatory process involving a segment or all of the extrahepatic biliary tree leading to loss of patency of the lumen and obstruction to bile flow. The result is cholestasis and chronic liver damage. With time, there is involvement of the intrahepatic biliary system. Uncorrected, the disease leads to complete biliary obstruction with cirrhosis. It is the most common cause of neonatal jaundice for which surgery is indicated and the most common indication for liver transplantation in children. BA is a progressive disease, and despite hepatoportoenterostomy at an “appropriate age,” 50 % of infants will require transplantation by 2 years of age and ultimately two-thirds will require liver transplantation in childhood. Although probably recognized since antiquity, the first major review of biliary atresia was provided by Thompson in 1891, who concluded that it represented inflammatory destruction of the bile ducts. Holmes discussed the possibility of bilioenteric anastomosis in 1916 and introduced the clinical classification into “correctable” and “non-correctable” types. The first successful surgery was reported by Ladd in 1928. For decades thereafter, confusion with and difficulties in distinguishing biliary atresia from other causes of infantile cholestasis and reports of “spontaneous cure” of biliary atresia and of harmful effects of surgery in patients with neonatal hepatitis resulted in delays in the timing of operative intervention with consequent dismal therapeutic results. Morito Kasai’s publication of success with portoenterostomy in these patients, in Japanese in 1959 and in English in 1968, not only represented a major therapeutic advance but also provided pathologists and investigators with invaluable hitherto unavailable specimens from the biliary tree.
10.5.2 Epidemiology
Biliary atresia occurs in 5–6 per 100,000 live births in North America and Europe, resulting in 250–400 new cases per year in the USA (Schreiber and Kleinman 2002). It is more common in East Asia, with a frequency of about 18 per 100,000 live births in Taiwan (Hsiao et al. 2008). In the USA, black females are >2 times as likely to give birth to an infant with BA than white females (The et al. 2007). The disorder has no significant hereditary component. Most twin studies have reported discordance for BA (Poovorawan et al. 1996; Silveira et al. 1991), and familial cases are exceptional (Kobayashi et al. 2008; Smith et al. 1991). Seasonal clustering has been noted, with a higher rate of BA observed in children born from December to April in one US study (Yoon et al. 1997), although no seasonal clustering was observed in a 20-year survey in Japan (Wada et al. 2007). The importance of early diagnosis and treatment has prompted searches for screening methods, the most successful of which to date has been the distribution of stool color charts in Taiwan and Japan to help parents identify abnormally pale stools. This appears to have led to earlier treatment and improved clinical outcome (Hsiao et al. 2008).
10.5.3 Clinical Features
Two major clinical forms of biliary atresia are recognized, but the BA phenotype may represent the final common pathway of several etiologies. The most common is the perinatal form which accounts for the majority of cases, and the other is the embryonic or fetal form. Children with the perinatal form are typically healthy appearing and of average weight at birth and pass normally pigmented stools. Between 4 and 8 weeks of age, cholestatic jaundice develops. The less common embryonic or fetal form occurs in 10–20 % of cases. Most of these children have the biliary atresia splenic malformation (BASM) syndrome, defined as a splenic malformation (polysplenia or asplenia) occurring in association with at least another major malformation, typically within the spectrum of laterality disorders (Table 10.4). In a retrospective multicenter study of 289 infants enrolled into the Childhood Liver Disease Research and Education Network, 242 (84 %) had isolated BA without a major malformation, 17 (6 %) had a major malformation but without a laterality defect, and 30 (10 %) were syndromic with laterality defects (Schwarz et al. 2013). Infants with BASM tend to come to clinical attention earlier than those without anomalies, but whether that is because of earlier onset of biliary obstruction and jaundice or because earlier evaluation is triggered by the associated anomalies is unclear. Despite earlier treatment, infants with BASM have a less favorable clinical outcome, a consistent finding observed in several studies (Chardot et al. 1999b; Davenport et al. 2006; Shneider et al. 2006). Though liver injury has been postulated to occur earlier in the embryonic than in the perinatal form, the observation that conjugated hyperbilirubinemia is present within the first 48 h of life in infants who develop the perinatal form of BA suggests that liver injury likely occurs very early after birth in all forms of BA (Harpavat et al. 2011).
Table 10.4
Congenital malformations most frequently associated with BASM
Spleen |
Polysplenia |
Asplenia |
Thoracic and/or abdominal situs inversus |
Dextrocardia |
Bronchial isomerism |
Midline symmetric liver |
Intestinal malrotation |
Interruption of the suprarenal segment of the inferior vena cava with azygous continuation |
Anomalous pulmonary venous drainage |
Preduodenal portal vein |
The more recently described cystic BA likely represents a separate category. Many of these cases are detected by antenatal ultrasound when cystic dilatation of the biliary tree suggesting a choledochal cyst is observed (Fig. 10.2). These infants typically present with jaundice at birth and no other major anomalies, in contrast with cases of BASM (Caponcelli et al. 2008; De Matos et al. 2005). Obstruction at the porta hepatis observed at laparotomy and histologic confirmation of a fibroinflammatory obstructive process distinguishes these cases as cystic forms of BA rather than obstructed choledochal cysts. These infants appear to have a more favorable outcome, and histologic liver damage at the time of portoenterostomy appears to be less extensive than most cases of BA (Caponcelli et al. 2008). In a large single-center retrospective review, cystic BA accounted for about 10 % of total cases of BA (Caponcelli et al. 2008).
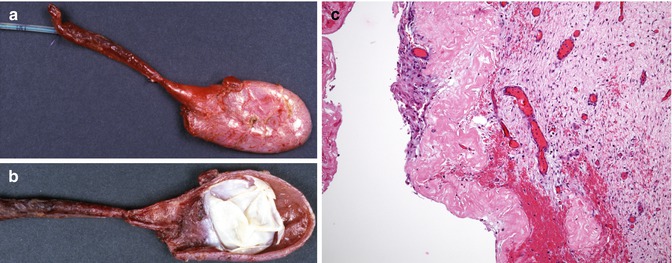
Fig. 10.2
Cystic biliary atresia. A cystic mass was identified by sonography at 36 weeks gestational age. The baby was delivered at 40 weeks’ gestation. Complete obstruction was noted at the porta hepatis and the infant underwent Kasai portoenterostomy at 10 days of age. (a) Dilated blunt-ending common bile duct, with absence of its distal portion, and cystic duct with small gallbladder, through the dome of which a catheter is present. (b) The mucosal surface of the opened common bile duct is covered by a thick white membrane. (c) Section through the dilated common bile duct. The epithelium is degenerated and there is extensive hyalinization of the subepithelial portion of the wall
10.5.4 Pathophysiology of Biliary Atresia
The pathogenesis of BA remains a mystery, and the relative infrequency of the disease in any one center makes investigation of large numbers of cases difficult. Most of the causal theories and research to date can be divided into several areas (Table 10.5 ).
Table 10.5
Proposed etiologies of biliary atresia
Viral infection |
Environmental toxin |
Immune dysregulation |
Autoimmunity |
Defects in morphogenesis |
Genetic predisposition |
Vascular abnormalities |
10.5.4.1 Viral/Toxin
Reports of seasonal clustering, the absence of a significant hereditary component and the occurrence of this disorder exclusively within the first 3 months of life have led to the suggestion that BA is the result of a disordered immune response to an initial viral or environmental insult. Reported outbreaks of an epidemic of BA among lambs in 1964 and 1988 by Harper et al. (1990) further substantiated the notion of a causative environmental factor. In each time period, there was a correlation with a drought and a change of grazing patterns by the pregnant ewes. In 1964, 60 of a flock of 400 died and in 1988 300 lambs died. Autopsies revealed an enlarged, firm, dark liver with a shrunken fibrotic gallbladder. No specific infectious or toxic agent has been identified to date.
A range of hepatotropic viruses have been demonstrated by PCR studies in about 50 % of the liver samples of BA patients obtained at the time of portoenterostomy, including CMV, reovirus, adenovirus, and enterovirus, with the presence of a viral infection-associated protein detected in an even larger proportion of samples (Rauschenfels et al. 2009). Three different viruses have been the focus of the most interest: reovirus, rotavirus, and cytomegalovirus. A relatively high prevalence of antibodies to reovirus type 3 was detected in infants with BA (Glaser et al. 1984) and neonatal hepatitis (Morecki et al. 1984a). Reovirus particles have been reported in human bile duct remnants by immunohistochemistry and electron microscopy (Morecki et al. 1984b), though this finding has been disputed (Bangaru et al. 1980). The presence of reovirus RNA was demonstrated in 55 % of frozen tissues of patients with BA (except, interestingly, those with associated polysplenia) as well as in 78 % of specimens from patients with choledochal cysts (versus 21 % of specimens from other hepatobiliary diseases) (Tyler et al. 1998). However, a similar search using archived paraffin-embedded material was negative (Steele et al. 1995). Intraperitoneal injections of reovirus in weanling mice induce hepatitis and inflammatory damage to intra- and extrahepatic bile ducts but without the development of the fully developed obstructive changes as observed in infants with biliary atresia (Szavay et al. 2002).
Riepenhoff-Talty et al. reported evidence of Rotavirus type C in liver tissues of patients with BA (Riepenhoff-Talty et al. 1996), though Bobo et al. could find no evidence for Rotavirus A, B, or C in their hepatobiliary samples (Bobo et al. 1997). Infection of newborn mice (but not the pregnant females) with Rotavirus type A (RRV) has produced complete biliary obstruction (Riepenhoff-Talty et al. 1993); pregnant mice that are immunized against RRV protect their newborns from developing RRV-induced cholestasis and biliary obstruction (Czech-Schmidt et al. 2001). Despite conflicting evidence of its significance in the etiology of human BA, RRV-induced murine BA has provided important insights into the role of cellular and humoral immunity in this disease (Mack et al. 2006).
Cytomegalovirus had been proposed as an etiologic agent in a number of early studies (Tarr et al. 1996; Hart et al. 1991). Fischler reported a higher prevalence of CMV antibodies in the mothers of infants with BA and higher CMV-IgM levels with greater amounts of IgG deposited in the hepatocyte membrane of affected infants (Fischler et al. 2005). Earlier studies looking for CMV DNA by PCR have yielded conflicting reports, some positive (Fischler et al. 1998) and others negative (Jevon and Dimmick 1999). More recently, Xu et al. analyzed liver tissue obtained at portoenterostomy by real-time PCR and found that 60 % were positive for CMV, in association with immunohistochemical detection of CMV antigen in the PCR-positive samples (Xu et al. 2012). Another recent study found evidence of an elevated liver T cell response to CMV in cases of BA, perhaps related to a developmental deficiency of T regulatory cells in the neonate (Brindley et al. 2012).
Other viruses implicated by PCR analysis in patients with biliary atresia and other cholestatic disorders have included herpes, adenovirus, and Epstein-Barr virus, either singly or in combination with cytomegalovirus (Domiati-Saad et al. 2000; Xu et al. 2012). Balistreri could find no serologic evidence for hepatitis B infection in neonates with BA or their mothers (Balistreri et al. 1980), and there appears to be no evidence for a role for hepatitis C in this disease (A-Kader et al. 1994). Drut and co-workers have reported evidence for human papillomavirus (HPV) types 6 and 18 by nested PCR in archived tissue from patients with BA and with neonatal hepatitis (Drut et al. 1998), though these findings could not be duplicated by others (Domiati-Saad et al. 2000). The variability of the data can be explained partly by differences in technique and sample size, by the frequent occurrence of many of these infections in neonates, and by the likelihood that infection by a variety of viruses during a critical window of time in a fetus or newborn could directly initiate damage or induce a poorly controlled inflammatory response that leads to continuing injury.
10.5.4.2 Immunologic/Autoimmune Dysfunction
Many studies support a role for immune dysfunction in BA, with much of this insight gained from the studies of previously mentioned murine models. The exclusive occurrence of the disease in the first few months of life further strengthens the notion of a developmental defect in the neonatal immune response. Although a viral infection may be the initial insult, other causes are possible. For example, the observation of the presence of circulating maternal CD8+ T cells (microchimerism) has also suggested the possibility of an alloimmune reaction as the initial trigger (Muraji et al. 2008). At present, the evidence supports dysregulation in a number of aspects of the native and adaptive immune cascade including antigen expression and presentation, T cell and Kupffer cell activation, cytokine release, apoptosis, and dampening of the immune response (Feldman and Mack 2012). As the first line of defense against infection, the innate immune system plays a pivotal role in the neonate in whom the adaptive response is not fully developed. That system includes macrophages, dendritic cells, natural killer cells, and neutrophils. These cells possess two major classes of receptors: Toll-like receptors (TLR) and pattern-recognition receptors (PRR). Bile duct epithelial cells also express PRRs. Upregulation of TRLs has been reported in the livers of patients with BA as compared to controls and choledochal cysts (Huang et al. 2007; Saito et al. 2011). Aberrant expression of MHC class II antigens has been reported in bile ducts, inflammatory cells, and damaged hepatocytes in BA (Kobayashi et al. 1997). Increased numbers and size of CD68+ macrophages producing high levels of tumor necrosis factor (TNF-α) have been identified in the portal tracts of livers of BA patients at diagnosis (Mack et al. 2004), and increased numbers of macrophages in the portal tracts have been correlated with a worse outcome (Kobayashi et al. 1997). Activated CD4+ and CD8+ from the adaptive immune response are also found at the time of diagnosis in the portal tracts and secrete various cytokines such as IL-2 and IFN-γ (Davenport et al. 2001; Mack et al. 2004). NK cells populate the vicinity of injured biliary epithelium in infants and the bile ducts of newborn mice with RRV-induced BA (Shivakumar et al. 2009). Depletion of NK cells in the mouse model prevented biliary injury (Shivakumar et al. 2009). Upregulation of osteopontin was demonstrated using gene expression studies in the livers of patients with BA (Bezerra et al. 2002). Osteopontin (OPN) is a cytokine which is important in inflammatory cell recruitment and fibrosis. Whitington et al. found increased levels of OPN in bile duct epithelium from BA livers that correlated with the extent of fibrosis (Whitington et al. 2005). Induction of the immune response in association with a developmental paucity of Tregs in the immediate postnatal period would result in an inability to dampen the inflammatory response and may be the cause of ongoing damage (Miethke et al. 2010; Brindley et al. 2012). Narayanaswamy and colleagues have identified increased circulating levels of a number of cytokines in the plasma of patients with BA at the time of diagnosis, which increased in the months following portoenterostomy suggesting continuing damage (Narayanaswamy et al. 2007). Upregulation of the immune response could also result from deficient silencing of pro-inflammatory genes due to deficient DNA methylation. Inhibition of methylation of zebrafish larvae resulted in upregulation of IFN-γ pathway genes and biliary damage, which could be reversed by administration of glucocorticoids (Matthews et al. 2011). Hypomethylation has been observed in human BA bile duct cells using an antibody to methylcytosine (Matthews et al. 2011).
An autoimmune response has been suggested by a number of studies. Abnormal antigen expression resulting from an infection or other insult to the biliary epithelium could trigger an autoimmune attack leading to injury and obliteration. Mack and colleagues demonstrated that adoptive transfer of T cells from RRV-diseased mice into naive syngeneic SCID recipients resulted in bile duct-specific inflammation, in the absence of detectable virus, suggesting an inflammatory response to biliary neoantigens. Furthermore, periductal immunoglobulin deposits and serum antibodies reactive to bile duct epithelial protein were detected in RRV-diseased mice (Mack et al. 2006).
10.5.4.3 Genetic Factors
BA is not thought to be a heritable disorder, but it is likely that genetics is a factor, its early onset suggesting genetic susceptibility to an acquired insult. Several genes have been postulated to act as possible disease modifiers. Mutations of the human JAG1 gene, which encodes a ligand for the Notch signaling pathway and is the hallmark of Alagille syndrome, were identified by Kohsaka et al. in 9 of 102 cases of BA (Kohsaka et al. 2002). These cases did not show any clinical features of Alagille syndrome and were associated with a worse clinical outcome than cases of BA without JAG1 mutations. Because these cases evidenced increased interleukin-8 production, the authors postulated a role for the JAG1 gene in the control of the inflammatory response and suggested that mutations of JAG1 could represent an aggravating factor (Kohsaka et al. 2002). Heterozygosity for non-M alleles of alpha1-antitrypsin was reported more frequently in children with chronic liver disease, including BA, and was associated with lower age at transplantation in BA (Campbell et al. 2007). Large-scale high-throughput analyses such as single nucleotide polymorphism (SNP) arrays and genome-wide association studies (GWAS) will likely identify genes or families of genes of interest, particularly those associated with control of the inflammatory response (Leyva-Vega et al. 2010). A GWAS of 200 Chinese children identified a high prevalence of an SNP on chromosome 10q24, close to the loci for the genes ADD3 and XPNPEP1, the former coding for a membrane-associated protein involved in cell-cell communication and the latter coding for an aminopeptidase involved in the regulation of vasodilatation and the metabolism of inflammatory mediators (Garcia-Barcelo et al. 2010).
10.5.4.4 Defective Morphogenesis
The strongest evidence supporting a defect in morphogenesis applies especially to infants with BASM. In BASM, the associated anomalies and similarities with the inv mouse suggest that alteration in gene expression at a critical time of development accounts for the anomalies. The inversin mouse (Inv), which has either a deletion or a recessive insertional mutation in the proximal region of chromosome 4, was reported to exhibit a similar constellation of features with anomalous development of the hepatobiliary system, as well as anomalies of visceral organ symmetry (Shimadera et al. 2007). The human inversin gene has been mapped to chromosome 9q, though no mutation of that gene was detected in a series of cases with BA and laterality disorders (Schon et al. 2002). As disorders of laterality have been associated with genetic defects in ciliary morphogenesis (“ciliopathies”), recently observed abnormalities in ciliary morphology by confocal immunohistochemistry in cholangiocytes in infants with BA (regardless of the presence of laterality defects), as compared to normal and disease controls, provide a possible link to the ciliopathies (Chu et al. 2012). Along that vein, mutations in the PKHD1 gene have been identified in BA patients in association with decreased immunostaining for fibrocystin (Hartley et al. 2011).
10.5.4.5 Vascular Etiology
An ischemic insult to the biliary tree has been another proposed theory. The bile ducts receive their blood supply exclusively from the hepatic arterial circulation. Interruptions of this flow account for bile duct damage in liver transplantation, as well as in a fetal sheep model (Pickett and Briggs 1969). Ultrasonographic and histologic abnormalities of the hepatic artery branches have been demonstrated histologically in BA patients (Ho et al. 1993). Dos Santos et al. observed progressive thickening of the hepatic arteries in BA which was associated with progressive loss of interlobular bile ducts (dos Santos et al. 2005).
10.5.5 Histopathology of Biliary Atresia
10.5.5.1 The Liver Biopsy in the Diagnosis of Biliary Atresia
The liver biopsy is a cornerstone of the diagnostic work-up of infants with cholestatic jaundice, and it is standard practice in most pediatric centers to obtain a percutaneous liver biopsy before surgical intervention (Moyer et al. 2004; Shneider et al. 2006). The broad range of disorders that can cause neonatal cholestasis makes interpretation of these biopsies challenging. The pathologist’s main role lies in recognizing histologic features of biliary obstruction, and BA is by far the most frequent cause of biliary obstruction in the neonate and infant. The assessment of a number of key histologic features yields the critical information needed to conclude that obstruction is present, provided that an adequate liver biopsy is performed at the right time. On the basis of consensus among pathologists participating in a recent multi-institutional study, an adequate biopsy should be a minimum of 2.0 cm long and 0.2 mm wide and not fragmented and ideally contain 10 portal areas (Russo et al. 2011). If it is a surgical wedge, it should be sufficiently deep to include six complete portal tracts independent of the liver capsule. Adequate clinical information should be available to the pathologist at the time of interpretation of the biopsy, including the patient’s age, the liver enzyme profile (particularly the level of gamma-glutamyltransferase, γGGT), the serum level of α1-antitrypsin, and any history of prior total parenteral nutrition (TPN). It is important to bear in mind that histologic features of BA may evolve over time. The earliest histologic changes of BA may be relatively nonspecific, such that biopsies performed too early in the course of the disease might result in a falsely negative diagnosis (Azar et al. 2002) (Fig. 10.3).
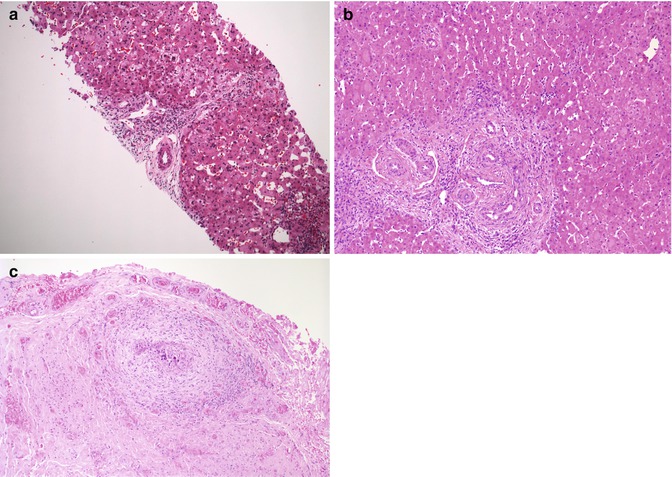
Fig. 10.3
(a) Liver biopsy in a 21-day-old neonate reveals mild portal expansion without significant bile duct proliferation. A cluster of myelopoietic cells is noted at the bottom of the picture. (b) At 40 days of age, the patient underwent exploratory laparotomy because of continued suspicion of biliary atresia. Portal expansion and bile duct proliferation are noted on the wedge biopsy. (c) The porta hepatis is obstructed
Relatively few studies, most based in single institutions and involving a limited number of cases, have evaluated the accuracy of liver biopsies in jaundiced infants (Brough and Bernstein 1974; Hays et al. 1967; Manolaki et al. 1983). Results were variable, the accuracy rate ranging from 65 to >90 %. In a recent multi-institutional study by the Biliary Atresia Research Consortium (BARC, now the Childhood Liver Disease Research and Education Network, or ChiLDREN), pathologists consistently recognized features of obstruction in approximately 90 % of clinically proven cases of BA (Russo et al. 2011). These features included portal expansion with proliferation of bile ducts and ductules (ductular reaction) and the presence of bile “plugs” in bile ducts, each of which had reasonably good interrater agreement in that study (Fig. 10.4 ). Canalicular and hepatocellular cholestases are usually present. Bile duct proliferation with bile plugs is the most specific finding for biliary obstruction and the one with the strongest discriminating value (Zerbini et al. 1997). Neutrophils are frequently present around the proliferating ductules and are part of the ductular reaction (Russo et al. 2011; Gouw et al. 2011) (Fig. 10.4b). When large clusters of neutrophils are present or if neutrophils are observed within the lumen of the ducts, a superimposed cholangitis should be suspected. Other than neutrophils, portal tracts may also contain cellular infiltrates of varying severity, most of which represents myelopoiesis (Fig. 10.4c). These histologic features contrast with those of neonatal hepatitis, in which variable degrees of lobular cholestasis are noted in association with other changes such as hepatocellular giant cell transformation and extramedullary hematopoiesis, with little or no changes in portal tracts or bile ducts. Nonetheless, variable degrees of extramedullary hematopoiesis, ballooning, and giant cell transformation of hepatocytes may also be observed in BA (Fig. 10.4d). Another feature observed with some frequency in cases of BA is the ductal plate configuration or “malformation” (DPM), characterized by curvilinear biliary structures arranged in a concentric pattern around a fibrovascular core within the portal tract mesenchyme (Russo et al. 2011) (Fig. 10.5 ). The persistence of this developmental configuration of the fetal intrahepatic biliary tree has led to the suggestion that DPM could be a marker for abnormal morphogenesis in BASM and, by extension, a predictor of a worse clinical outcome (Low et al. 2001; Shimadera et al. 2008). However, this histologic feature has poor interobserver agreement (Russo et al. 2011). Moreover, recent studies have found no relationship between the presence of DPM in liver biopsies and an association with BASM (Davenport et al. 2006; Pacheco et al. 2009).
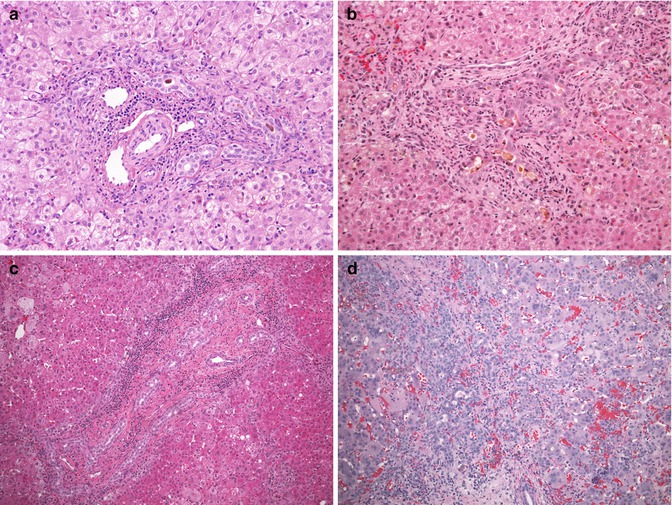
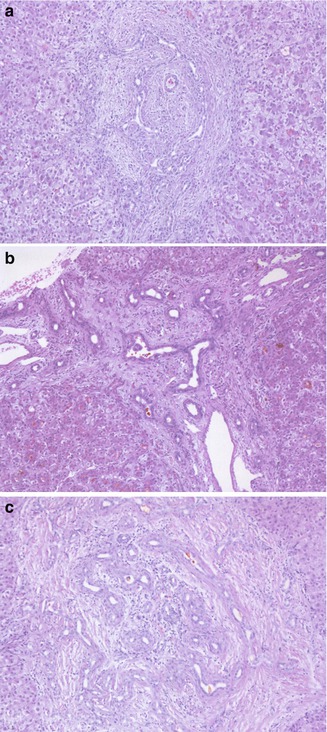
Fig. 10.4
Liver biopsy findings in biliary atresia. (a) The liver biopsy in this 7-week-old infant reveals an expanded portal tract, with bile duct and ductular proliferation and bile plugs within the biliary lumen. A mild cellular infiltrate representing myelopoiesis is present within the portal space. The surrounding lobule is unremarkable. (b) A liver biopsy in this 6-week-old infant demonstrates a marked periductal neutrophilic infiltrate raising the possibility of a cholangitis. An atretic biliary tree and porta hepatis were confirmed at laparotomy. (c) A significant portal cellular infiltrate representing myelopoiesis can be observed in cases of BA. (d) Unusually marked lobular changes and giant cell transformation of hepatocytes are noted in the biopsy from this 10-week-old infant
Fig. 10.5
Cases of biliary atresia with “ductal plate malformation”. (a) Wedge liver biopsy from a 10-week-old child at the time of the Kasai portoenterostomy. No other malformation was present in this patient. (b) Wedge liver biopsy at the time of Kasai portoenterostomy in a 2-month-old with polysplenia and heterotaxia. (c) Wedge liver biopsy at time of Kasai operation from a 3-month-old patient with a combination of biliary atresia and long chain fatty acyl-CoA dehydrogenase deficiency
Although histologic features of obstruction should lead to consideration of surgical exploration, other diseases enter into the differential diagnosis of “obstructive cholestasis” and BA (Table 10.6 ). Infants with α-1 antitrypsin deficiency may present with liver dysfunction and cholestasis. In a large nationwide prospective study carried out in Sweden, 14 of 127 infants with the PIZZ phenotype identified from 200,000 screened newborns had prolonged obstructive jaundice (Sveger 1976). The histologic features of liver biopsies in these infants can be indistinguishable from those of BA, and normal levels of serum α-1 antitrypsin should always be documented prior to surgical exploration in an infant with obstructive cholestasis on a liver biopsy (Fig. 10.6a). In the neonate, PAS-D-positive intracytoplasmic globules are generally not as prominent as in the older patient. Hypoplasia of the extrahepatic ducts, defined as abnormally small but patent ducts and usually associated with syndromic paucity of intrahepatic bile ducts (Alagille syndrome), may result in confusion with BA because of clinical and radiographic evidence of biliary obstruction (Maurage et al. 1991; Kaye et al. 2010) (Fig. 10.6b). The constellation of clinical features of Alagille syndrome should orient the clinician. Though liver biopsies in Alagille syndrome typically reveal bile duct paucity, bile duct proliferation suggesting obstruction can be noted early in the evolution of the disease (Deutsch et al. 2001; Emerick et al. 1999). Confusion with BA has led to portoenterostomies being performed in some cases of Alagille syndrome, with an adverse effect on the clinical outcome of these patients (Kaye et al. 2010). Cystic fibrosis may also present as neonatal cholestasis in about 1 % of cases. The liver biopsy features in CF patients with neonatal cholestasis, presumably representing a selection of more clinically worrisome cases, can closely overlap with those of biliary atresia, and cholangiography may be required to exclude it (Fig. 10.6c) (Shapira et al. 1999; Perkins et al. 1985). Characteristic inspissation of pink or orange material in bile ducts may be focal or even absent. In most reported cases, jaundice typically clears within 1 year without evidence of progression to chronic liver disease (Shapira et al. 1999). Some of these children have associated conditions (α-1 antitrypsin deficiency, hypopituitarism, total parenteral nutrition) that enhance the risk of cholestasis (Lykavieris et al. 1996; Shapira et al. 1999). Of note, patients with BA and concomitant cystic fibrosis have been described with a hypoplastic extrahepatic tree (Greenholz et al. 1997). Liver biopsies in reported cases of neonatal sclerosing cholangitis associated with ichthyosis (NISCS) due to mutations in the gene coding for Claudin 1 on chromosome 3q27-q28 may show portal fibrosis with ductular proliferation and thus mimic BA (Hadj-Rabia et al. 2004). Operative cholangiography demonstrating patent extrahepatic ducts but abnormal intrahepatic ducts, in association with the constellation of ichthyosis, scarring alopecia, and other dysmorphic features, distinguishes these cases from BA (Hadj-Rabia et al. 2004).
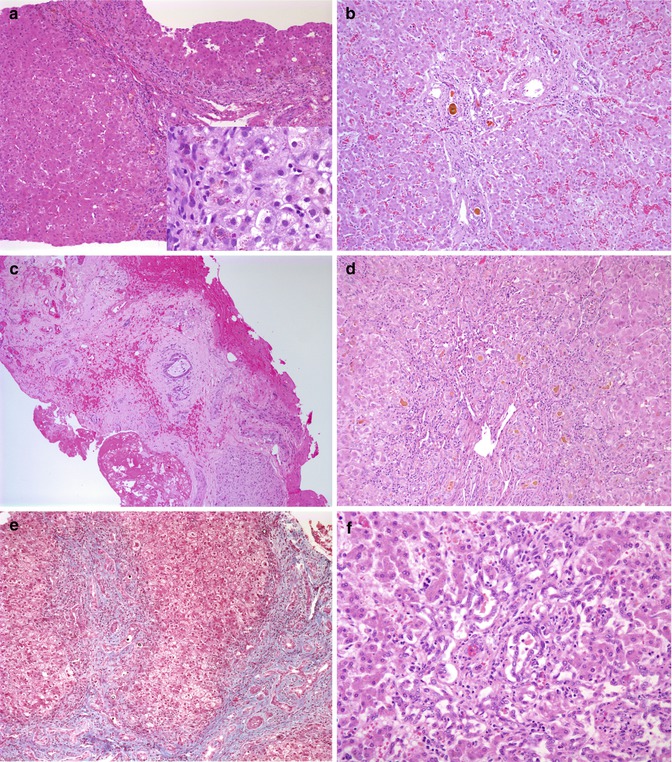
Table 10.6
Differential diagnosis of disorders with features of obstruction on a liver biopsy and helpful distinguishing features
Alpha-1-antitrypsin deficiency | Low or absent serum alpha-1-antitrypsin level, PiZZ phenotype, PAS-D-positive granules generally not prominent in liver biopsies in neonates |
Choledochal cyst | Ultrasonographic and cholangiographic findings |
Alagille syndrome (early) | Associated malformations, mutation in JAG1 gene |
Cystic fibrosis | History of meconium ileus, failure to thrive, positive sweat test |
TPN hepatopathy | History of prematurity and TPN use |
Neonatal sclerosing cholangitis | Ichthyosis, scarring alopecia, dysmorphism, patent extrahepatic but abnormal intrahepatic ducts on cholangiogram |
Progressive familial cholestasis type 3, ABCB4 disease | Usually older infants at presentation; absence of immunohistochemical staining for MDR3 along canaliculi |
North American Indian cirrhosis | Incidence restricted to Native American groups in Canada |
Tumor, stone, or other mechanical obstruction |
Fig. 10.6
Biliary proliferation in disease other than biliary atresia. (a) Bile ductular proliferation in a liver biopsy from a 10-week-old infant with cholestasis. Further investigation revealed alpha-1-antitrypsin deficiency with a PIZZ phenotype. Small poorly distinguished intracytoplasmic globules are noted with the PAS stain following diastase (inset). (b) Autopsy liver from a 4-month-old boy with Alagille syndrome features portal expansion and bile duct proliferation with bile plugs. (c) Hypoplastic biliary tree from a 4-month-old with Alagille syndrome. (d) Portal expansion, bile duct proliferation, and inspissated bile in a liver biopsy from a 4-month-old with cystic fibrosis. (e) A liver biopsy from a 2-month-old Cree boy with North American Indian childhood cirrhosis reveals extensive portal fibrosis with bile duct and ductular proliferation with bile “plugs.” Despite the obvious similarity to an obstructive disorder, the extrahepatic biliary tree and gallbladder were normal. (f) Portal expansion and bile duct proliferation in a liver biopsy from a 42-day-old child with cholestasis subsequent to total parenteral nutrition since birth
In addition to its role in diagnosis, evaluation of the liver biopsy might also reveal prognostically significant histologic features, such as the extent of fibrosis, that might help predict outcome after Kasai portoenterostomy. However, studies assessing the correlation of histologic features in the liver biopsy with clinical outcomes post Kasai portoenterostomy have so far produced conflicting results, some suggesting that fibrosis portends a poor outcome (Weerasooriya et al. 2004; Pape et al. 2009), whereas others have emphasized the significance of lobular changes as predictors of clinical outcome (Azarow et al. 1997). A gene expression profile study carried out at Cincinnati Children’s Hospital found that children with a liver biopsy displaying mainly fibrosis had a molecular profile distinct from those with predominantly inflammatory appearance and that the fibrotic “signature” was strongly associated with a worse clinical outcome (Moyer et al. 2010).
10.5.5.2 Anatomy and Histopathology of the Resected Segment of the Biliary Tree
The destructive inflammatory process that underlies BA may involve a short segment of a duct, an entire duct, or the entire system. Many different classifications of BA have been proposed over the years, based on the anatomy of the atretic segment and correlated with surgical “correctibility” or “non-correctibility” as regards the bilioenteric anastomosis. All essentially recognize three broad types. The most comprehensive classification is that used in the Japanese Biliary Atresia Registry which includes the following main types: type I, atresia of the common bile duct; type II, atresia of the hepatic ducts; and type III, atresia at the porta hepatis. The latter accounts for about 90 % of cases (Davenport 2012). Each main type is then further subclassified, according to this classification, as to the appearance of the distal and proximal portions of the remnant (Fig. 10.7)
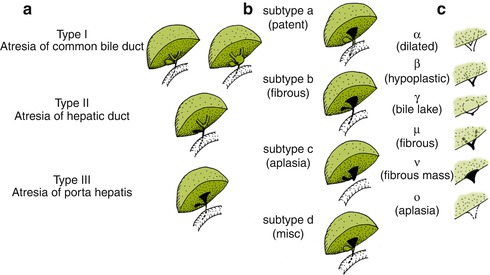
Fig. 10.7
Ohi classification of biliary atresia remnants into three main types (a), with distal (b) subtypes and proximal (c) subtypes (Reprinted from Superina et al. (2011))
Histologic study of the resected biliary remnant has been described by numerous investigators (Chandra and Altman 1978; Gautier and Eliot 1981; Matsuo et al. 1984; Miyano et al. 1977; Tan et al. 1994). Gautier and Eliot (1981) classified the remnants into three types: type 1, an absent lumen replaced by a fibrous cord; type 2, multiple small lumina, usually less than 50 μm, arrayed around a fibroinflammatory cord; and type 3, presence of a central lumen, sometimes containing bile and corresponding to an altered bile duct (Fig. 10.8 ). When a central lumen is present, extensive degenerative changes and injury to the epithelium are usually observed, with inflammation and fibrosis of the surrounding wall. Squamous metaplasia of the biliary ductules may be observed (Stahlschmidt et al. 2008). The most active site of inflammation is usually the porta hepatis, with fibrous obliteration of the distal biliary tree. Alternating foci of patency and obliteration are most unusual, in our experience. Occasionally, nodules of cartilage have been noted at the porta (Mirkin and Knisely 1997; Stahlschmidt et al. 2008). The gallbladder in Ohi type II and III forms of atresia is often atrophic or collapsed, with epithelial degeneration and fibrosis of the wall. Multiple small lumina may be seen (Fig. 10.8d). It may be patent and distended in type I.
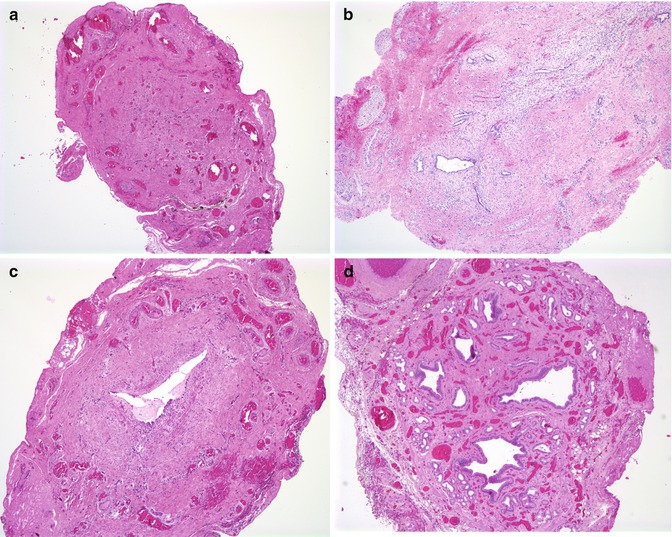
Fig. 10.8
Features of biliary remnants observed at portoenterostomy. (a) A fibrous cord without any apparent epithelial remnant in a 6-week-old child. (b) Small diminutive biliary structures surrounded by concentric fibrosis are noted in this section from a 2-month-old child. (c) Bile duct with single major lumen with periductal inflammation and fibrosis and extensive epithelial degenerative changes from a 6-week-old child. (d) The gallbladder can be atrophic and have a complex internal structure characterized by multiple small lumens surrounded by muscular tissue
A number of studies have attempted to correlate the size of bile duct remnants at the porta hepatis with the outcome of the portoenterostomy. Chandra and Altman found that optimal biliary drainage was achieved with a diameter of the proximal biliary remnant greater than 150 μm (Chandra and Altman 1978). Matsuo et al. found that the sum of the areas of the different bile duct lumens in a section of the porta hepatis was perhaps a better indicator than the largest diameter (Matsuo et al. 1984). However, Tan and colleagues determined that the degree of liver damage at the time of surgery was the best correlate with outcome (Tan et al. 1994). A study by Langenburg showed no correlation of outcome with bile duct size (Langenburg et al. 2000). A more recent study found that an increased maximal length measured in any direction for the residual bile ducts, as well as the numbers of bile ducts at the porta, correlated with a better clinical outcome (Mirza et al. 2009). The difficulty in obtaining constant and reproducible sections permitting accurate measurements is a major methodological problem inherent with most of these studies, and prognostication based on the size of the remnants up to now appears to have little practical value.
10.5.5.3 Clinical Outcome Post-portoenterostomy
Biliary cirrhosis develops within 6 months in untreated cases and, eventually, in most children treated by portoenterostomy. Nevertheless, most clinical studies support the notion that BA patients who undergo surgery prior to 90 days of age have a better outcome (Davenport 2012; Serinet et al. 2009; Superina et al. 2011). A review of portoenterostomies from 1980 until 1995 at King’s College Hospital showed that more than 50 % of children were alive with their native livers at 5 years and 40 % at 10 years if the hepatoportoenterostomy was performed by 100 days of life (Davenport et al. 1997). Whether surgery at a much earlier age (<30 or 40 days) can actually improve the current outcomes remains controversial. The presence of BASM is clearly associated with a worse clinical outcome in most series (Davenport 2012; Shneider et al. 2006). More limited forms of BA (Ohi type I) appear to correlate with a better prognosis (Davenport 2012; Superina et al. 2011) as does the cystic form of BA. Clinical evidence of successful biliary drainage 3 months post-portoenterostomy, defined as total bilirubin less than 2 mg/dL, was associated with a lower risk of transplant or death, according to one recent study (Superina et al. 2011). Several studies from different countries have emphasized the rather sensible notion that the experience of the operating team is an important determinant of success (Balistreri et al. 1996; Chardot et al. 1999a; McKiernan et al. 2000). This has led to an effort to centralize the care and treatment of these children in some European countries and has spurred the establishment of multi-institutional collaboration in the USA through the ChiLDREN. Centralization of the treatment of cases of BA to three institutions in the UK appears to have improved clinical outcomes there (Davenport et al. 2011).
10.5.5.4 Liver Pathology Post-portoenterostomy
Liver specimens following portoenterostomy are most frequently obtained for evaluation of persistent jaundice and recurrent fevers or from hepatectomy specimens obtained at transplantation or autopsy. Hepatectomy specimens from jaundiced infants or children with BA are typically coarsely nodular and green (Fig. 10.9a, b). Bile pigment in hepatocytes and canaliculi; ballooning of hepatocytes and giant cell transformation; periportal hepatocellular accumulation of copper, the latter detectable using the rhodanine stain; and variable presence of Mallory’s hyaline characterize findings in the lobule. It has long been recognized that cells with a myofibroblastic phenotype produce fibrosis in BA (Lamireau et al. 1999; Ramm et al. 1998). These cells have been demonstrated to accumulate around proliferating ductules (de Freitas et al. 1986). Ductular epithelial cells also appear to elaborate fibrosis-related cytokines and matrix-remodeling proteins such as metalloproteinases (Al-Masri et al. 2006; Faiz Kabir Uddin Ahmed et al. 2000) and have been proposed to undergo differentiation into myofibroblastic cells (epithelial to mesenchymal transition, EMT) (Diaz et al. 2008), perhaps by upregulating the Hedgehog pathway (Omenetti et al. 2011), although lineage tracing studies have challenged this concept (Chu et al. 2011).
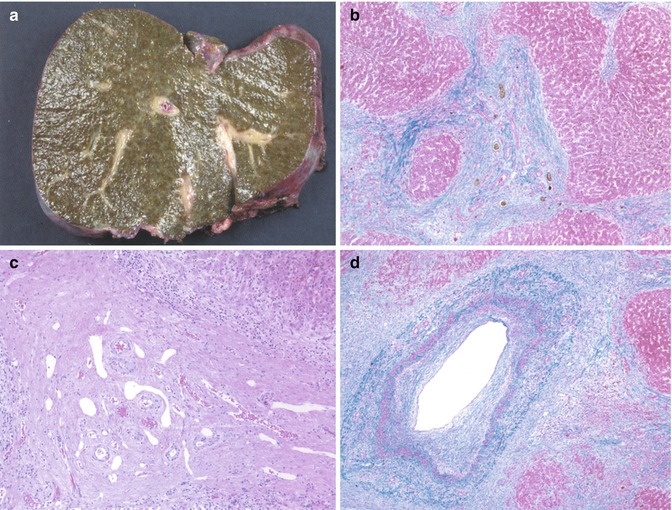
Fig. 10.9
Chronic changes in biliary atresia. (a) Recipient hepatectomy specimen at transplantation, 18 months after Kasai portoenterostomy. The liver surface is coarsely nodular and green tinged. (b) Low-power section from the liver in figure “A” reveals cirrhosis with wide fibrous bands and proliferating bile ducts with inspissated bile. (c) Bile duct paucity in transplant specimen, 18 months post Kasai portoenterostomy. (d) Sclerotic portal vein noted in transplant specimen 11 months post Kasai portoenterostomy
Progressive loss of intrahepatic bile ducts is noted (Fig. 10.9c), in untreated cases, as early as 5–6 months, and occurs in most patients despite treatment, even when adequate biliary drainage is established (Nietgen et al. 1992). In our experience and that of others, this is primarily seen in patients who survive longer with their native livers post-portoenterostomy and may be asymmetrical in its distribution (Nietgen et al. 1992). Injury by bile stasis, peribiliary ischemia, and continuation of the original insult may all be contributing factors. Heterogeneity and unevenness in the distribution of bile duct loss and occasional nodules of better preserved parenchyma may reflect local differences in biliary obstruction or fibrosis (Hussein et al. 2005; Ijiri et al. 2001). Concomitant sclerosis and atrophy of the portal venous system has been noted by 3-D reconstruction (Ohuchi et al. 1986) and may contribute to portal hypertension (Fig. 10.9d).
Cysts filled with dense inspissated bile, so-called bile lakes, develop in up to 20 % of children after portoenterostomy, usually near the hilum (Tainaka et al. 2007; Watanabe et al. 2007). Histologically, most consist of extravasated bile surrounded by a fibroinflammatory reaction. Residual epithelium is observed in a minority of cases (Fig. 10.10 ). There seems to be no communication between these cavities and the intrahepatic biliary tree (Fonkalsrud and Arima 1975), although rare cases have been reported to represent concomitant Caroli’s disease (Takahashi et al. 1997). Cholangitis occurs in up to 50–90 % of cases, usually within 3 months, most often due to gram-negative bacteria (Ecoffey et al. 1987), but can also occur years after surgery. Bile lakes may also be found in the cirrhotic livers of anicteric children with BA. Most studies suggest they worsen the patient’s prognosis. Surgical resection or percutaneous drainage has been advocated in some cases (Inoue et al. 2000).
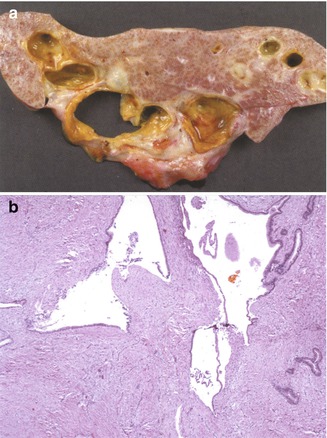
Fig. 10.10
Biliary cysts following Kasai portoenterostomy. (a) Large bile-filled cysts are noted in the hepatectomy specimen of this 15-month-old child, 1 year post Kasai portoenterostomy. Liver function was preserved, but the child required continuous antibiotic treatment for repeated infections. (b) Low-power microscopic view of the large, dilated biliary structures noted in (a)
Malignancies have been reported in a small percentage of children with biliary atresia, mainly hepatocellular carcinoma (Hadzic et al. 2011) but also hepatoblastoma (Tatekawa et al. 2001) and cholangiocarcinoma (Kulkarni and Beatty 1977). Focal nodular hyperplasia has also been reported (Ohtomo et al. 1991; Okugawa et al. 2008). These tumors can occur many years after the initial surgery and underscore the need for careful long-term follow-up.
10.6 Choledochal Cysts
10.6.1 Epidemiology and Classification
Choledochal cysts represent a segmental dilatation of the biliary ductal system and a surgically treatable cause of direct hyperbilirubinemia. They are believed to be congenital, though patients can present in infancy, later in childhood, and as adults (Rha et al. 1996; Suita et al. 1999). Acquired, progressive development of a choledochal cyst has also been observed (Han et al. 1997). The frequency of choledochal cysts is approximately 1:150,000 live births in Western countries and much higher in Asian countries (Singham et al. 2009). There is a marked (4:1) female preponderance, regardless of race (Singham et al. 2009). The most widely used anatomic classification is the Todani modification of the Alonso-Lej classification, which proposes five main variants of which type 1 is the most common, accounting for greater than 90 % of cases (Singham et al. 2009; Todani et al. 1977). The variants are illustrated in Fig. 10.11 . Concomitant involvement of the intrahepatic bile ducts is reportedly frequent with dilatation of the extrahepatic tree, occurring in more than 20 % of cases, according to Todani (Singham et al. 2009; Todani et al. 1977). Some authors consider type 5 to be essentially synonymous with Caroli’s disease (Singham et al. 2009). This is disputed by others who contend that Caroli’s disease, and Caroli’s syndrome when associated with congenital hepatic fibrosis, is defined by heritability and an association with renal anomalies and thus has a different etiology than choledochal cysts (D’Agata et al. 1994).
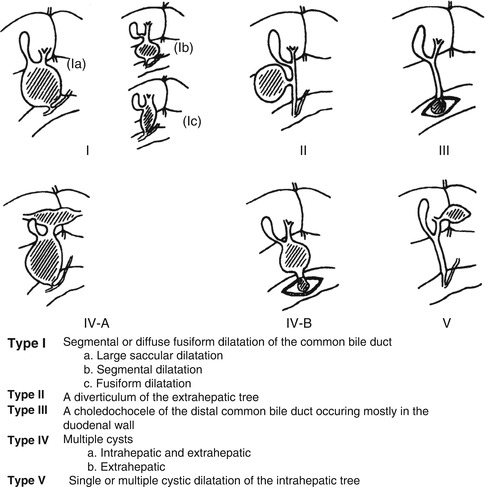
Fig. 10.11
Anatomic classification of choledochal cysts
10.6.2 Pathogenesis
Theories of pathogenesis of choledochal cysts primarily include bile reflux and obstruction of the distal biliary tree (Tanaka 1995). However, the various forms of dilatation of the bile ducts may not necessarily share a common cause. An anomalous junction between the common bile duct and the pancreatic duct, well outside the muscle of the ampulla of Vater, with a long common channel is observed in a majority of patients with choledochal cysts (Babbitt 1969; Todani et al. 1984). An abnormal pancreatobiliary duct junction was identified by a prospective study in up to 4 % of Korean patients undergoing ERCP; these same authors detected this anomaly by magnetic resonance imaging and cholangiography in up to 80 % of children diagnosed with a choledochal cyst (Kim et al. 2002). This is believed to result in reflux of bile containing pancreatic enzymes (Jona et al. 1979; Komi et al. 1984). Reflux of bile has also been postulated as a risk factor for carcinoma of the biliary tree (Baumann et al. 1987; Chijiiwa et al. 1993), as well as carcinoma developing in those individuals with an abnormal pancreaticobiliary junction without a choledochal cyst (Suzuki et al. 1999). Ectopic location of the papilla of Vater in the more distal duodenum, also resulting in a longer common biliary channel, has been reported by Li et al. in a study of patients with congenital biliary dilatation (Li et al. 2001). However, reflux alone cannot explain the dilatation observed in those cases diagnosed antenatally, some as early as 15–16 weeks’ gestation, before significant fetal pancreatic enzyme secretion. Obliteration of the distal bile duct has been reported in association with and as a cause of a choledochal cyst diagnosed antenatally (Tsang et al. 1994).
10.6.3 Clinical Presentation and Diagnosis
The classic presentation is the triad of pain, abdominal mass, and jaundice. More commonly, the triad is not present and one or more of the signs or symptoms are present. In one study, 53 % of children with a choledochal cyst presented with an abdominal mass, compared to 21 % of adults (Huang et al. 2010). There can be associated serious consequences including recurrent pancreatitis, biliary cirrhosis, portal hypertension (the cyst impinges the portal vein), and biliary carcinoma (vide infra). Perforation of the cyst can occur leading to bile peritonitis. Alternatively, the findings can also be very subtle. There can be intermittent pain or jaundice. In the most subtle case, the choledochal cyst is simply an incidental radiologic finding. The cyst is usually visible on ultrasound. More than two-thirds of patients have an elevated GGT and approximately half have elevations of bilirubin. The most useful diagnostic study is ultrasound. Further delineation of the extent and location of cysts is aided by endoscopic retrograde cholangiopancreatography (ERCP) or MR cholangiopancreatography. HIDA scans can differentiate choledochal cysts from biliary atresia, as contrast will be visualized in the small bowel with a choledochal cyst, but not with biliary atresia (Goldman and Pranikoff 2011).
10.6.4 Pathology
In most cases, histologic sections reveal a thick-walled structure composed of dense connective tissue with interspersed smooth muscle, frequently without an epithelial lining and usually little to no inflammation (Fig. 10.12). Variable lengths of a flattened epithelium, focal or extensive ulcerations, and stone formation may also be observed. Cystic BA can be differentiated from a choledochal cyst by evidence of fibroinflammatory obliteration of the biliary tree in the former. Mucosal hyperplasia (Imazu et al. 2001) and papillomatosis of the biliary tree (Ohita et al. 1993) have been reported in choledochal cysts. Examination of the liver in virtually all cases reveals varying degrees of bile duct proliferation, cholestasis, and inflammation. Fibrosis and cirrhosis are noted in a significant proportion of cases (Nambirajan et al. 2000). Some studies have suggested that liver damage is more severe in infancy (Suita et al. 1999).
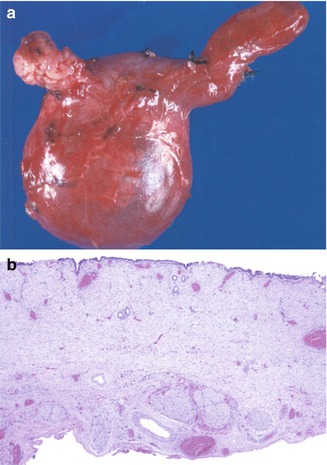
Fig. 10.12
Choledochal cyst. (a) Resection of biliary tree with a choledochal cyst in a 4-month-old girl. The gallbladder dome is toward the upper right hand portion of the picture. (b) Low-power view microscopic view of cyst wall reveals a simplified, flattened, focally absent epithelium and a thick fibrous wall
10.6.5 Malignancy
A number of malignancies have been associated with choledochal cysts. The most common is adenocarcinoma (cholangiocarcinoma) (Goldman and Pranikoff 2011; Kamisawa et al. 2008), although rhabdomyosarcoma has also been observed (Patil et al. 1992). Multiple adenocarcinomas may arise, in either a synchronous or metachronous fashion (Yoshikane et al. 1998). The overall risk of developing cholangiocarcinoma with choledochal cysts is approximately 10 % (Chapman 1999), varying from 2 to 28 % (Fieber and Nance 1997). The risk is much greater in patients presenting over the age of 20 than in those who present in infancy (Goldman and Pranikoff 2011) (Fig. 10.13). It has been reported in patients as young as 12 years (Imazu et al. 2001). The incidence has been estimated to be 0.7 % in the first decade of life, 6.8 % in the second decade, and 14.3 % in successive decades (Rall and Chung 1995). The risk of carcinoma in the residual portion of the tract, including the intrahepatic portion, remains elevated even after surgical excision (Fujisaki et al. 1999).
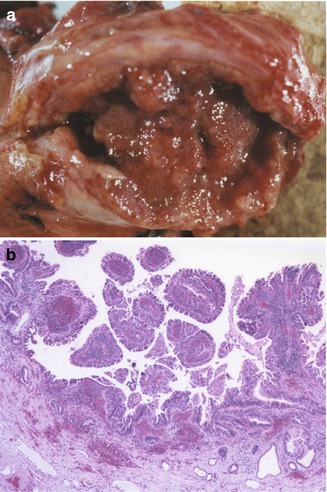
Fig. 10.13
Cholangiocarcinoma arising in a choledochal cyst. (a) Resected specimen from a 27-year-old female reveals a fleshy hemorrhagic lesion filling the cyst cavity and infiltrating the wall. (b) The cystic cavity is diffusely lined by a dysplastic epithelium forming papillary folds, with infiltration of the wall (gross specimen and microscopic slides courtesy of Dr. Emma Furth, Hospital University of Pennsylvania)
10.6.6 Treatment
Cholecystectomy with cyst excision and choledochojejunostomy is the treatment of choice. Hepaticoduodenostomy has also been proposed, although increased complications such as gastritis and esophagitis have been reported with this procedure and appear to be greater in children (Goldman and Pranikoff 2011). Early exploration is recommended in those cases diagnosed antenatally, because of the possibility of biliary atresia. Regardless of the surgical intervention, long-term follow-up is required because of the continued risk for development of malignancy in the residual bile duct or at the anastomotic site.
10.7 Sclerosing Cholangitis
Primary sclerosing cholangitis (SC) is a chronic inflammatory disorder of unknown etiology characterized by inflammation, fibrosis, and stricturing of the intrahepatic and/or extrahepatic biliary tree. Secondary sclerosing cholangitis refers to disorders in which bile duct damage is mediated by another underlying disorder (lithiasis, infection, Langerhans cell histiocytosis). Though relatively infrequent in children, PSC is nevertheless an important cause of pediatric chronic liver disease, accounting for about 3 % of liver transplants performed in children annually in the USA (Shneider 2012). Published experience in pediatrics is limited by comparison with the literature in adult patients, but several large pediatric series emphasize the heterogeneous nature of the disease in children (Batres et al. 2005; Debray et al. 1994; Feldstein et al. 2003; Mieli-Vergani and Vergani 2001; Miloh et al. 2009; Wilschanski et al. 1995). In addition, inherited disorders such as progressive familial intrahepatic cholestasis type III, due to a mutation in the gene ABCB4, may have been included in some of these series because of its clinical similarity. Several clinical forms which appear particular to children have also been recognized more recently and are described later in this section: neonatal sclerosing cholangitis and sclerosing cholangitis with autoimmune features (autoimmune sclerosing cholangitis).
10.7.1 Idiopathic (Primary) Sclerosing Cholangitis (PSC)
The exact prevalence of PSC in the pediatric population is unknown. It is probably fair to state that primary sclerosing cholangitis is underestimated and, in many instances, unrecognized in children. As in adults, there is a close association with chronic inflammatory bowel disease, with predominance of ulcerative colitis (Batres et al. 2005). Conversely, PSC has been reported to occur in fewer than 10 % of patients with IBD (Dotson et al. 2010).
10.7.1.1 Pathogenesis
Although the precise pathogenesis remains unknown, several areas have become targets of interest (reviewed in Krones et al. 2012). The close relationship with IBD has led to the concept of the “leaky gut” in which a disrupted intestinal barrier leads to circulating inflammatory cells which can cross-react with extraintestinal epitopes and result in damage. Studies in mice have also demonstrated that modulation of the composition of bile (“toxic bile”) could induce bile duct injury. Thus, Abcb4/Mdr2 mice, the rodent ortholog of ABCB4/MDR3, which show absence of phosphatidylcholine in bile, develop sclerosing cholangitis (Fickert et al. 2004). Genetics also clearly plays a role, as first-degree relatives of patients with PSC are at a higher risk of developing PSC and IBD (Bergquist et al. 2008). Genome-wide association studies (GWAS) have revealed a number of genes of interest involving inflammation, cholangiocyte function, fibrogenesis, and cancerogenesis (Naess et al. 2012). Interestingly, there appears to be a discrepancy between the loci of susceptibility observed for IBD and those for PSC (Krones et al. 2012). Functional impairment of the cystic fibrosis transmembrane receptor (CFTR, mutated in cystic fibrosis), resulting in impaired bicarbonate secretion at the cholangiolar surface, has been postulated to cause damage to the bile ducts by allowing uncontrolled membrane permeation of protonated glycine-conjugated bile acids (Beuers et al. 2010). CFTR chloride channel function was found to be impaired in children with PSC (Pall et al. 2007).
10.7.1.2 Autoimmune Sclerosing Cholangitis
Sclerosing cholangitis in children is frequently associated with autoimmune hepatitis (AIH), either simultaneously or within a short interval, and the term autoimmune hepatitis/sclerosing cholangitis overlap syndrome (or just overlap syndrome for short) and its equivalent, autoimmune sclerosing cholangitis (AISC), have been used to refer to these patients. AISC in children has the same prevalence as AIH type 1 and is more frequent than in adults (Mieli-Vergani and Vergani 2010). Fifty-five children with serologic (positive autoantibodies, high IgG) and histologic (interface hepatitis) features of AIH followed prospectively for 16 years underwent a cholangiogram at presentation. Approximately half the patients had cholangiographic abnormalities consistent with PSC. In a quarter of the cases, liver histology did not show characteristic biliary involvement despite the cholangiographic features (Gregorio et al. 2001). Virtually all ASC patients were positive for ANA and/or SMA. Nearly half the cases of ASC in that series were associated with inflammatory bowel disease, and pANCA could be detected in three-quarters. Eight of these patients showed a progressive cholangiopathy on repeat liver biopsies. The diagnosis of AISC is based on demonstrating characteristic cholangiographic features in a child with liver disease and the presence of autoimmune antibodies. Because distinguishing between ASC and AIH may be clinically impossible, it has been suggested that magnetic resonance cholangiopancreatography (MRCP) be performed systematically in cases of AIH.
10.7.1.3 Diagnosis
Children with PSC are often asymptomatic at the time of presentation even though they might have advanced disease, making clinical recognition difficult. The earliest symptoms are fatigue and pruritus. Later signs include fevers, chills, night sweats, and right upper quadrant pain. These latter findings are only present in 10–15 % at presentation. Jaundice usually reflects advanced disease or superimposed biliary infection. The serologic findings are consistent with cholestatic disease. The serum alkaline phosphatase and gamma-glutamyltransferase are generally elevated, whereas ALT is usually less than 300 IU/L. The bilirubin may fluctuate due to transient blockage of strictured bile ducts by sludge or stones. Persistent elevation indicates advanced disease. Additional supportive lab findings include hypergammaglobulinemia and pANCA (30–80 %). A variety of causes of chronic liver disease in children which can mimic PSC, such as cystic fibrosis and ABCB4 disease, should also be excluded (Shneider 2012).The diagnosis is made by finding characteristic multifocal stricturing on cholangiography. MR cholangiography is helpful and as the technology improves may someday replace conventional ERCP. Strictures can occur in any part of the biliary tree. The strictures may be short with intervening normal sections or may be long. Eighty-seven percent of patients have a combination of intra- and extrahepatic stricturing. A small percent may have isolated intra- or extrahepatic stricturing (Lee and Kaplan 1995). The gallbladder and cystic duct may also be involved.
10.7.1.4 Pathology
A percutaneous liver biopsy can support the diagnosis but is rarely diagnostic. The earliest changes are frequently characterized by a “cholangitic” picture, with bile duct and ductular proliferation and a portal inflammatory infiltrate including lymphocytes, plasma cells, and neutrophils (Fig. 10.14a) (Batres et al. 2005). Despite an appearance suggestive of obstruction, bile pigment is usually inconspicuous early on. Bile duct epithelium frequently shows degenerative changes. A marked lymphocytic infiltrate with interface activity is typically observed in early biopsies of cases of “overlap” syndrome, or autoimmune cholangitis, resulting in an appearance essentially indistinguishable from autoimmune hepatitis (Fig. 10.14b). The presence of significant cholangiographic biliary changes should lead to the proper diagnosis (Boberg et al. 1996). Increasing portal fibrosis, bridging, and, eventually, cirrhosis are evidence of further disease progression. The characteristic “onion-skin” fibro-obliterative process (Fig. 10.15 ) surrounding bile ducts is noted only in a minority of cases in early biopsies. This is probably because of sampling, due, at least initially, to the focal nature of the process and by the size of the ducts involved. The fibrosis is frequently associated with epithelial degeneration and atrophy of the bile duct.
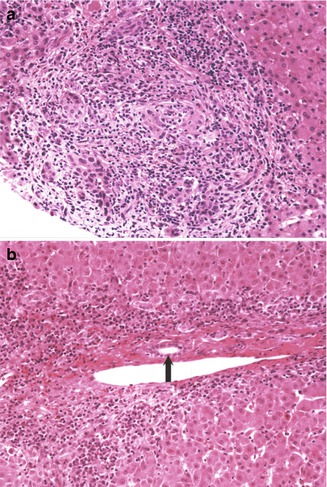
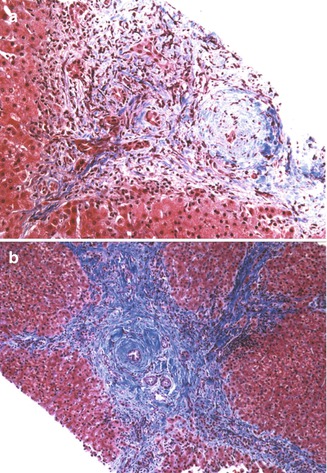
Fig. 10.14
Primary sclerosing cholangitis. (a) Initial liver biopsy of a 17-year-old male with ulcerative colitis and elevated liver enzymes reveals a “cholangitic” appearance with bile duct proliferation and a mixed inflammatory infiltrate including numerous neutrophils. (b) Initial liver biopsy in a 12-year-old male with ulcerative colitis is suggestive of autoimmune hepatitis with a lymphoplasmacellular infiltrate and interface hepatitis. A bile duct is indicated by the arrow
Fig. 10.15
Primary sclerosing cholangitis. (a) Liver biopsy reveals expanded portal tracts with fibrosis, bile ductular proliferation, and a small shrunken biliary remnant surrounded by a concentric dense collar of fibrosis (Masson trichrome stain). (b) Bridging portal fibrosis and “onion skinning” around bile ducts (Masson trichrome stain)
Ludwig has divided the histologic changes into four stages (Ludwig 1989); though used in some comparative studies, histologic staging for prognostication is limited by sampling variability and by the occurrence of different stages simultaneously in the same biopsy. In our experience, most children present with histologically advanced disease, mirroring the experience of Floreani and colleagues, who found that children with PSC tended to present with a greater severity of clinical and histologic activity than adults (Floreani et al. 1999b).
Hepatectomy specimens obtained at transplantation typically reveal micronodular cirrhosis or bridging portal fibrosis. Active inflammatory destruction of the larger biliary structures can be observed in many specimens (Fig. 10.16a). Obliterated bile ducts replaced by a concentric scar and lobular cholestasis are characteristic late features (Fig. 10.16b).
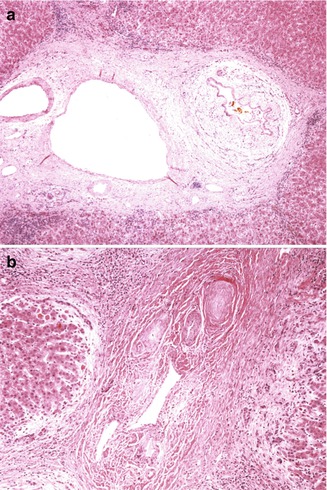
Fig. 10.16
Primary sclerosing cholangitis (hepatectomy specimens). (a) Low-power view of large portal tract reveals connective fibrosis and inflammation of a bile duct. (b) Cords of dense acellular fibrous tissue have replaced the biliary structures, resulting in bile duct paucity
The term small-duct PSC has been proposed to define PSC affecting bile ducts that are too small for cholangiographic identification (Ludwig 1991) and is essentially synonymous with the older term “pericholangitis,” which referred to biliary abnormalities noted in liver biopsies of patients with chronic inflammatory bowel disease (IBD). Diagnosis rests on liver histologic findings consistent with PSC, a normal cholangiogram, and elimination of secondary causes of sclerosing cholangitis (Singal et al. 2011). It appears to follow a more indolent course than large duct PSC (Singal et al. 2011). Its incidence in children is unknown.
10.7.1.5 Prognosis and Treatment
Medical management for PSC rests essentially on the prevention of treatment of complications of chronic liver disease. Medical management is similar to that recommended for adults, although the risk of cholangiocarcinoma appears to be less (Shneider 2012). Liver transplantation is a successful treatment modality for advance liver disease. Recurrence of the disease in the transplant has been described in up to 30 % of patients (Fig. 10.17) (Batres et al. 2005; Feldstein et al. 2003).
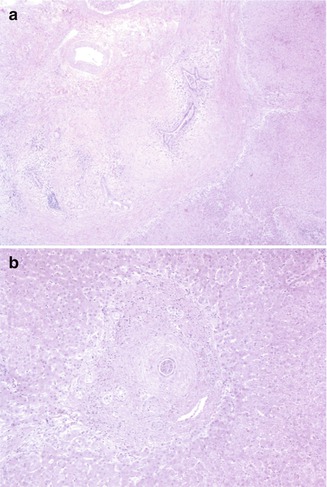
Fig. 10.17
Primary sclerosing cholangitis – recurrence in allografts. (a) Low-power microscopic view of hilar area of resected allograft reveals inflammatory destruction and concentric fibrosis around biliary structures, as noted in the original native hepatectomy specimen. (b) Concentric periductular paucicellular fibrosis is noted in this allograft
10.7.2 Neonatal Sclerosing Cholangitis
Amedee-Manesme et al. in 1987 described eight children with cholestasis from the first week of life, followed by early cirrhosis and portal hypertension (Amedee-Manesme et al. 1987). Debray et al.’s series of 56 children with sclerosing cholangitis included 15 with a neonatal onset (Debray et al. 1994). Many of these cases have been linked to a mutation in the claudin-1 (CLDN1) gene on chromosome 3q27-q28 and referred to as the neonatal ichthyosis-sclerosing cholangitis (NISCH) syndrome (Baala et al. 2002; Hadj-Rabia et al. 2004). These neonates present with cholestasis of variable severity in the first weeks of life, usually with elevated GGT levels. They are associated with prominent extrahepatic clinical manifestations, mainly ichthyosis and scarring alopecia. Additional features of this disorder include enamel hypoplasia, oligodontia, and hypodontia (Paganelli et al. 2011). It is postulated that a defect in claudin-1 in hepatocytes and cholangiocytes leads to loss of cell polarity and to paracellular leakage of bile. Likewise, a severe impairment of epidermal barrier function is postulated to cause dehydration of the skin and ichthyosis (Paganelli et al. 2011). Liver histology can be indistinguishable from biliary atresia, although a range of abnormal histologic findings has been noted, from mild to severe, including bile duct paucity (Amedee-Manesme et al. 1987; Nagtzaam et al. 2010; Paganelli et al. 2011). The typical “onion-skin” appearance of periductal fibrosis is not appreciated in these cases. Operative cholangiography has revealed patency of the extrahepatic bile ducts, in contrast to biliary atresia, in association with thin and irregular-appearing extra- and intrahepatic bile ducts (Girard et al. 2012). The clinical course is variable, and there is no genotype-phenotype correlation. Although jaundice usually subsides during the first year of life, progression to biliary cirrhosis appears inevitable (Girard et al. 2012).
10.7.3 Secondary Sclerosing Cholangitis
Secondary causes of sclerosing cholangitis include entities as diverse as infections in association with primary and secondary immunodeficiencies and infiltrative processes such as Langerhans cell histiocytosis, choledocholithiasis, and ischemia resulting from toxic injury to the biliary vascular plexus due to floxuridine injection (Table 10.7). Langerhans cell histiocytosis is one of the more frequent causes of secondary sclerosing cholangitis in the pediatric population (Debray et al. 1994) (Fig. 10.18). The characteristic Langerhans cells may not be detected in the liver biopsies, despite evidence of biliary damage (Braier et al. 2002), and the diagnosis then rests on the presence of a concomitant infiltrate in other sites such as skin and bones.
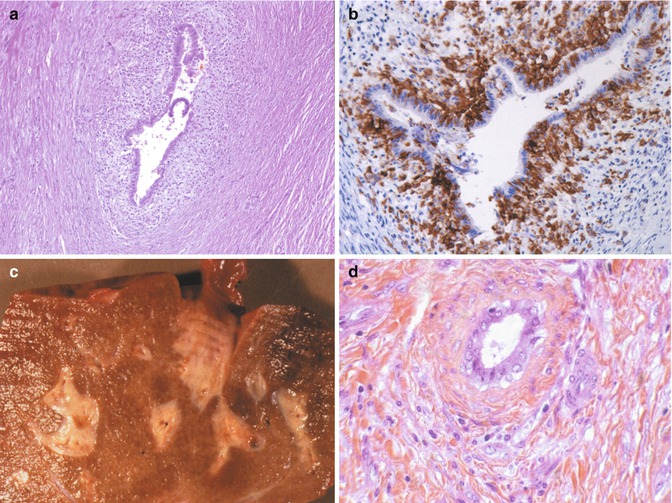
Table 10.7
Secondary causes of sclerosing cholangitis in children
Immunodeficiencies |
(a) Primary immunodeficiency |
Hyper-IgM syndrome |
(b) AIDS-associated cholangiopathy |
(c) Post-liver transplantation |
Cryptosporidial infection |
Langerhans cell histiocytosis |
Choledocholithiasis |
(a) Sickle cell disease |
(b) Total parenteral nutrition |
Hodgkin’s disease |
Cystic fibrosis |
Post-biliary surgery |
Ischemia (floxuridine) |
Sclerosing cholangitis in association with genetic syndromes |
(a) Neonatal ichthyosis-sclerosing cholangitis syndrome |
(b) Kabuki syndrome |
(c) Laurence-Moon-Bardet-Biedl syndrome |
Fig. 10.18
Secondary sclerosing cholangitis due to Langerhans cell histiocytosis. (a) Liver specimen from a 3-year-old child reveals active destruction of the larger bile ducts by an infiltrate consisting of Langerhans cells. (b) CD1a immunohistochemical staining highlights the infiltrating Langerhans cells within the wall of a bile duct. (c) Autopsy liver from a 4-year-old child with Langerhans cell histiocytosis is nodular with prominent, expanded, and fibrotic portal areas. (d) High-power view showing concentric periductular fibrosis. No residual Langerhans cells were noted in this liver
10.8 Stricture of the Extrahepatic Ducts
Strictures can be a rare cause of obstructive jaundice in children. Biliary strictures occur most commonly in adults as a consequence of trauma, previous surgeries, chronic pancreatitis, neoplasia, and cholelithiasis (Bonnel et al. 2001).They have been reported following surgery, such as hepatic transplantation, probably resulting from ischemic damage to the bile ducts (Campbell et al. 1994; Garcia-Gallont et al. 1999; Peclet et al. 1994) and repair of a choledochal cyst in an adult (Dunn et al. 1984); a postsurgical traumatic neuroma causing stricture has been observed (Mentha et al. 1999). In one reported series of 7 cases ranging in age from 2.5 to 15 years at presentation, one child had been treated with chemo- and radiotherapy for leukemia, whereas the obstruction was idiopathic in the remainder. The strictures were noted at different levels of the biliary tree, without a specific site of predilection. Histopathological examination of the strictures showed collapsed biliary structures with fibrosis, inflammation, and epithelial degeneration, reminiscent of sclerosing cholangitis. Changes in the liver ranged from obstructive features to cirrhosis (Bowles et al. 2001). Strictures have been observed in infants following repair of gastroschisis (Hancock et al. 1989). They have been reported as a complication of chemo- and radiotherapy for malignancies (Hughes et al. 1994). Idiopathic or congenital strictures in childhood are described most commonly at the junction of the hepatic ducts. On occasion they are found at the ampulla of Vater and thought to be secondary to pancreatitis. If the lesion is distal, a sphincteroplasty is the treatment of choice. More proximal strictures are treated with choledochoenterostomy.
10.9 Perforation of Bile Ducts
Spontaneous perforation of the bile ducts is a rare condition which tends to occur within the first few months of life but can occur later in childhood (Niedbala et al. 2000) and even in adults (Paladugu et al. 1998). Since first described in 1932 by Dijkstra, approximately 150 cases have been reported (Evans et al. 2010; Pereira et al. 2012). The most common site of perforation is the junction of the common bile duct with the cystic duct (Hasegawa et al. 2000). The usual presentation is that of an infant with jaundice, acholic stools, and bilious ascites (Banani et al. 1993). A sealed-off biliary abscess can result in local symptoms such as biliary obstruction (Chardot et al. 1996) or can mimic an intestinal cyst (Goldberg et al. 2000) or gastric outlet obstruction (Kumar et al. 2001). It can constitute a medical emergency with abdominal distension and shock. It has been diagnosed in the antenatal period, where it may be a cause of fetal ascites (Chilukuri et al. 1990).
The pathogenesis is not understood, but it is likely multifactorial. Proposed theories include a congenital weakness of the bile duct wall, distal obstruction, and anomalous union of the pancreaticobiliary ductal system (AUPBD) (Evans et al. 2010; Sai Prasad et al. 2006). In support of the latter theory, Ando reported 13 cases of spontaneous perforation in 187 children with choledochal cysts (Ando et al. 1998). An ischemic etiology is also suggested by a report of biliary perforation with necrotizing enterocolitis (Ibanez et al. 1999). Spontaneous perforation in infancy has been observed to lead to biliary atresia (Davenport et al. 1996). Perforation of bile ducts has also been described with stenosis of the ampulla of Vater (Donahoe and Hendren 1976), with acalculous cholecystitis (Shah et al. 1990), and following endoscopic retrograde pancreatography (Martin and Tweedle 1990). Birth trauma may be a factor in some cases (Topuzlu Tekant et al. 1994).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


