Texas Children’s Hospital
Consultations
HB
85
185
HCC
33
57
Fibrolamellar carcinoma
5
6
Cholangiocarcinoma
2
1
Undifferentiated sarcoma
18
31
Rhabdomyosarcoma
2
3
Mesenchymal hamartoma
14
26
Hemangioma
15
5
Angiosarcoma
4
7
Rhabdoid tumor
2
22
Nested stromal–epithelial tumor
0
5
Total
180
348
Great progress in the treatment of HB and some sarcomas has been achieved in the past quarter century. This progress can be attributed in part to improved imaging and anesthesia and to a growing cadre of surgeons familiar with hepatic anatomy thanks to orthotopic transplantation and the use of living donors. Mostly, the benefits have derived from cis-platinum-based chemotherapy. With the advances has come the recognition that thorough histopathologic characterization of HB has prognostic significance and should influence the choice of therapy. These advances have done less for disease that is all too often disseminated at presentation, including most hepatocellular carcinomas (HCC) and the small undifferentiated cell and rhabdoid variants of HB.
Molecular genetic studies of hepatic tumors are providing considerable new information via modalities such as comparative genomic hybridization (CGH), CHIP and SNP (single nucleotide polymorphisms) copy number variation, and whole genome sequencing. Patterns of gene expression ultimately will permit a better understanding of pathogenesis and will lead to therapies that avoid the severe toxicity of current regimens while more effectively attacking the neoplastic cells. But traditional morphologic diagnoses and complete surgical extirpation remain the mainstay of therapy. This chapter summarizes the current data on the antecedent and acquired genetic abnormalities in liver tumors while emphasizing those aspects of histopathology that are essential for diagnostic accuracy.
14.1 Etiology
14.1.1 Incidence and Epidemiology
The compilations by Dehner (1978), Weinberg and Finegold (1986), and Stocker (2001) incorporated data from 19 surveys, representing regions of Australia, Canada, Italy, Japan, Sweden, Switzerland, Taiwan, the United Kingdom, the United States, and the former USSR. The AFIP series is likely to be least selective and most representative of the US population. There were 24 cases per year over 30 years from 1970 to 1999, and when these are added to prior studies, the distribution of diagnoses is shown in Table 14.2. Sixty percent of the tumors arose from hepatocytes, of which 96 % were malignant, and 61 % arose from immature fetal or embryonal elements. Sixty-six percent of all tumors were malignant. Benign tumors, especially hemangiomas, and focal and diffuse nodular hyperplasia collectively contributed about 20 % of the total and probably are underreported, but they do contribute occasionally to difficulties in clinical and even pathological diagnoses.
Table 14.2
Primary liver tumors in children 1956–2001
Primary tumors | Number | Percent |
|---|---|---|
Hepatoblastoma | 737 | 37 |
Hepatocellular carcinoma | 422 | 21 |
Adenoma | 50 | 2.5 |
Focal nodular hyperplasia | 94 | 5 |
Benign vascular tumors | 290 | 15 |
Mesenchymal hamartoma | 133 | 7 |
Sarcoma (embryonal, angio, rhabdomysarcoma) | 156 | 8 |
Other | 90 | 4 |
Total | 1,972 | 100 |
14.1.2 Etiology
The causes of hepatocellular neoplasms include infection, constitutional and heritable metabolic diseases, and environmental or drug exposure (Table 14.3). Maternal smoking had a two times increased risk of HB not accounted for by birth weight in both New York (McLaughlin et al. 2006) and the United Kingdom (Pang et al. 2003). Neither parental infertility nor its treatment, including “assisted reproductive technology,” was associated with a greater incidence of HB among 383 mothers of affected infants in a recent COG study (Puumala et al. 2012). There was a significant excess of maternal exposure before and during pregnancy to metals used in welding and soldering, lubricating oils, and protective greases (Buckley et al. 1989). Paternal exposure to metals was also greater. An HB was found in a stillborn fetus at 23 weeks whose mother was an artist exposed to volatile hydrocarbons. Occasionally, patients with HB have had umbilical hernia, undescended testes, renal dysplasia, agenesis of the gallbladder, or other minor malformations.
Table 14.3
Precursors of hepatic neoplasia
Perinatal exposure |
Oral contraceptives |
Phenytoin |
Ethyl alcohol |
Neonatal factors |
Extreme prematurity |
Maternal hepatitis B carrier |
Metabolic disease |
Tyrosinemia |
Von Gierke’s disease, glycogenosis type 1a |
Glycogenosis III, IV, VI |
Niemann–Pick, type C |
Malformations |
Hemihypertrophy, Beckwith–Wiedemann syndrome |
Simpson–Golabi–Behmel syndrome |
Sotos syndrome |
Autosomal recessive polycystic disease |
Multiple hemangiomatosis |
Ataxia–telangiectasia |
Fanconi anemia |
Budd–Chiari syndrome |
Von Recklinghausen’s neurofibromatosis |
Biliary tract disease |
Extrahepatic atresia |
Familial cholestatic cirrhosis |
Alagille syndrome |
Parenteral alimentation |
Drugs |
Oral contraceptives |
Anabolic steroids |
14.1.3 Prematurity
The US National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) program includes approximately 14 % of the population, and it revealed an average annual percentage increase in the incidence of HB from 1973 to 2007 of 5.2 Howlander (2013). This change might be explained by HB occurring in surviving premature infants. Ikeda et al. (1997) first reported from Japan that HB accounted for 58 % of all malignancies in children who weighed less than 1,000 g at birth. Further analysis of the Japanese Children’s Cancer Registry data revealed that 15 of 303 HB (5 %) between 1985 and 1995 occurred in post-premature infants weighing less than 1,500 g. This rate was ten times greater than that for all live births. The relative risk for HB for children who weighed less than 1,000 g at birth was 56.9 versus 5.0 for those 1,000 g to 1,499 g versus 1.6 for 1,500 g to 2,499 g in New York State from 1985 to 2001 (McLaughlin et al. 2006).
Of 77 children with HB in the German registry, three (4 %) were premature infants who required parenteral nutrition, a treatment that has been lifesaving for many small premature infants but has been reported to lead to cirrhosis in many survivors and in two cases to HCC. It has not previously been associated with HB (Hadzic et al. 2011). Another 26-week premature infant developed HB 9 years later. Of patients in the current COG study for whom data are available, 13 % were born before 30 weeks and 16 % more before 35 weeks, so 29 % were premature. Spector and colleagues (2009) evaluated 17,672 childhood cancer diagnoses versus 57,966 randomly selected control children and confirmed the exceptionally high rate of very low birth weight for HB only. Maruyama et al. (1999) found no parental exposures to potential risk factors in the histories of 15 affected premature infants. Typical postnatal management was required for respiratory distress, and longer use of ventilators and oxygen was noted in those with a higher stage of HB. Similarly, there was more use of furosemide in those patients. Only one infant had hepatitis, due to cytomegalovirus, and one had cholelithiasis. Interestingly, 11 % of the premature US babies had normal α-fetoprotein (AFP) levels at the time of diagnosis versus 4 % for the whole HB population. The age of detection and histologic features of HB following prematurity are indistinguishable from other HB (Fig. 14.1).
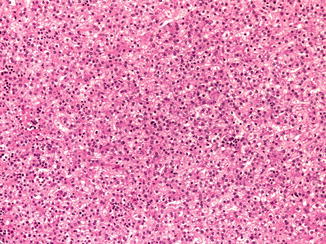
Fig. 14.1
Prematurity and hepatoblastoma. The patient was 3 years old at the time of diagnosis following birth at 23 weeks of gestation. The fetal cells are larger than normal fetal hepatocytes and have a higher nuclear/cytoplasmic ratio. The nuclei are regular and round. Clusters of normoblasts indicate extramedullary hematopoiesis as in the fetal liver. The cytoplasm of the hepatocytes varies from eosinophilic to clear depending on the amount of glycogen. Mitoses are not seen in this well-differentiated fetal HB
14.1.4 Hepatitis B Virus
The most common pathogenetic factor of hepatocellular cancer is hepatitis B virus (HBV), which has induced HCC in the sons of chronic carrier mothers in Asia as young as 4 years of age. With vaccination at birth, the incidence of HCC in childhood in Taiwan was decreased by 50 % from 1981–1986 to 1990–1994, and this decline has been confirmed in a 20-year follow-up (Chang et al. 2009). Out of 5.5 million vaccines at birth, only 64 HCC were found, only when mothers were HBe positive or inadequate doses of vaccine were given. Vaccination obviously will have enormous benefit in eradicating the chronic carrier state and preventing carcinoma in adults. Recent surveys, however, show an increase in HCC in the United States (http://seer.cancer.gov/csr). Hepatitis C virus is now responsible for the majority of HCC in Japan and other countries with excellent public health programs. Unlike hepatitis B, it has not been responsible for childhood HCC because it appears to act via chronic inflammation, with repeated episodes of injury and regeneration providing for accumulation of somatic mutations. When hepatitis B infection is acquired after the perinatal period, it also takes 20 or more years to be carcinogenic, but in the very young, whose livers are growing rapidly, it is capable of integrating into host DNA and producing neoplasia very quickly (Fig. 14.2) (Tanaka et al. 1986; Hino et al. 1984). Integration sites for HBV–DNA vary considerably, but some of them involve loci that may be relevant to the process of carcinogenesis, such as cyclin A and D1 (Nishida et al. 1994). The spectrum of chromosomal gains and losses demonstrable by CGH in HCC arising in patients with HBV infection differs from those in uninfected persons. Mutations in the viral genome itself may be important in carcinogenesis in adults; this has not been evaluated in children. Specific genetic features of the pre-S2 region of the HBV itself are correlated with carcinogenesis (Han et al. 2011).
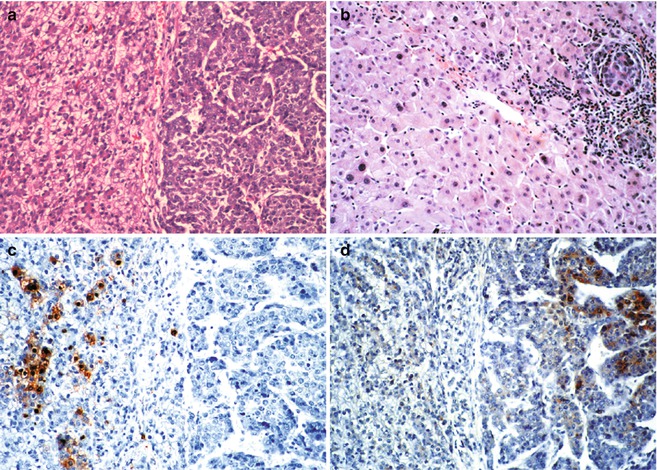
Fig. 14.2
Hepatoblastoma and hepatocarcinoma side-by-side in a 15-year-old male with perinatally acquired Hepatitis B virus infection. (a) Typical fetal hepatoblastoma cells on the left form cords of cells with relatively clear cytoplasm and small regular nuclei. There is modest mitotic activity. On the right are large macrotrabecular collections of hepatocytes that are relatively well differentiated. Mitotic activity is abundant. (b) Sections of the liver apart from the tumor show, in addition to intravascular spread of the hepatocarcinoma cells on the right, dysplastic changes in the nontumoral hepatocytes. Nuclei are pleomorphic and hyperchromatic. (c) Hepatitis B core antigen is abundant in the nuclei and cytoplasm of the hepatoblastoma cells on the left, but none is found in the hepatocarcinoma. (d) Contrarily, alpha fetoprotein is present in the hepatocarcinoma cells, but none is found within the fetal hepatoblastoma
14.1.5 Gender
The particular vulnerability of male children to get hepatocellular neoplasms is intriguing. It is true not only for perinatally acquired HBV infection but also for HCC arising in children with autosomal recessive metabolic disease, such as glycogen storage disease type 1, and for HB in families with familial polyposis (Giardiello et al. 1996). A partial explanation, from studies of experimental carcinogenesis, is that only male mice expressing the transforming growth factor TGFα gene developed HCC. TGFα is a strong stimulus to hepatocyte replication and normally is expressed much more in younger individuals. TGFα is produced by more than 65 % of HCC, providing for autocrine stimulation. It is also expressed strongly in fetal HB cells that have low proliferative activity (Kiss et al. 1998), supporting a paracrine role for this molecule. Disruption of the pRB/E2F pathway and inhibition of apoptosis were observed in TGFα and c-Myc transgenic mice (Santoni-Rugiu et al. 1998). An androgen receptor-regulated gene, cell-cycle-related kinase (CCRK), was found to upregulate beta-catenin/TCF signaling in HCC cells, and overexpression of CCRK was observed in the tumors of high stage and worse patient survival (Feng et al. 2011). Transgenic mice expressing RBMY (RNA-binding motif) in the liver develop hepatocellular neoplasia and this molecule regulates androgen receptor activity (Tsuei et al. 2011). It was detected in 36 % of human HCC and 4/6 HB but only in the tumors and only in males (Tsuei et al. 2004). The role of sex steroids in hepatocellular neoplasms is well known, primarily because of the frequent development of the adenoma in women who took oral contraceptives when they contained large quantities of estrogen. One study found fibrolamellar carcinoma in two young women exposed to estrogens (Malt et al. 1983), but, to our knowledge, this has not been observed subsequently. However, endogenous abnormalities in sex hormone metabolism may also contribute, as observed in two boys, aged 15 years and 17 years who failed to enter puberty, who had gynecomastia and developed fibrolamellar carcinomas having high levels of aromatase in the tumor (Agarwal et al. 1998). Exogenous androgens are also implicated, especially in patients with Fanconi anemia, most of whom have had benign adenomas (Fig. 14.3a), but HCC have also been observed (Fig. 14.3b).
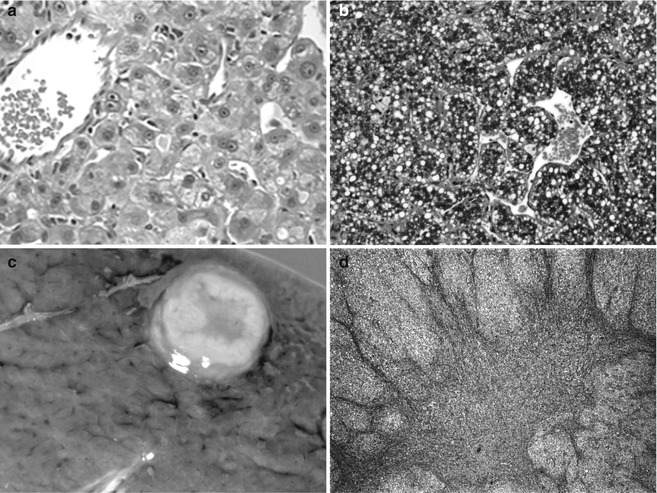
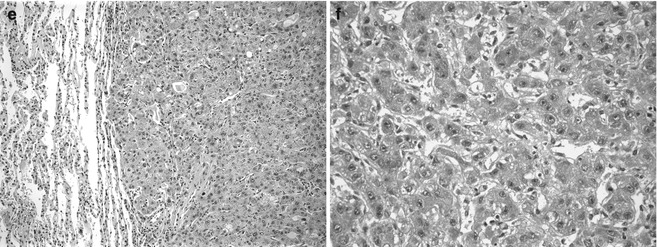
Fig. 14.3
Hepatocellular neoplasia in patients with underlying genetic disease. (a) This 12-year-old patient with an adenoma and Fanconi anemia had been treated with anabolic steroids. The adenoma cells are larger than normal hepatocytes and there is minimal nuclear pleomorphism. No bile ducts were present within the well-circumscribed lesion. (b) Hepatocellular carcinoma is growing in a macrotrabecular pattern with abundant cytoplasmic lipid. The cells are moderately well differentiated. This specimen comes from a 15-year-old girl with Fanconi anemia whose sister was similarly affected. (c) This well-circumscribed lesion is in the liver of a 16-year-old male with glycogen storage disease 1A. (d) The central depression is due to scarring and is consistent with focal nodular hyperplasia. (e) Metastatic hepatocellular carcinoma is shown in the lung of a 22-year-old man with glycogen storage disease type 1a. (f) Infants with hereditary tyrosinemia type 1 fail to cease AFP production due to persistent metabolic injury. This dysplastic liver was from a 4-month-old
14.1.6 Cholestatic Disease
Several chronic cholestatic syndromes have been precursors to HCC, including long-term use of parenteral nutrition, and extrahepatic biliary atresia (Hadzic et al. 2011). Of 109 orthotopic liver transplants in children reported from France, 3 had HCC, 1 of whom had extrahepatic biliary atresia and another Byler syndrome (PFIC1, FIC mutation). Adenomatous hyperplasia, which may be a preneoplastic lesion, was observed in 11 of 80 (14 %) biliary atresia livers. The genetic basis for neonatal giant cell hepatitis and other chronic cholestatic syndromes associated with HCC and cholangiocarcinoma (Fig. 14.4) is becoming better defined with progress in molecular genetics (see Chap. 11). However, the carcinomas in these cases have not been included in the many molecular and cytogenetic studies of HCC described below.
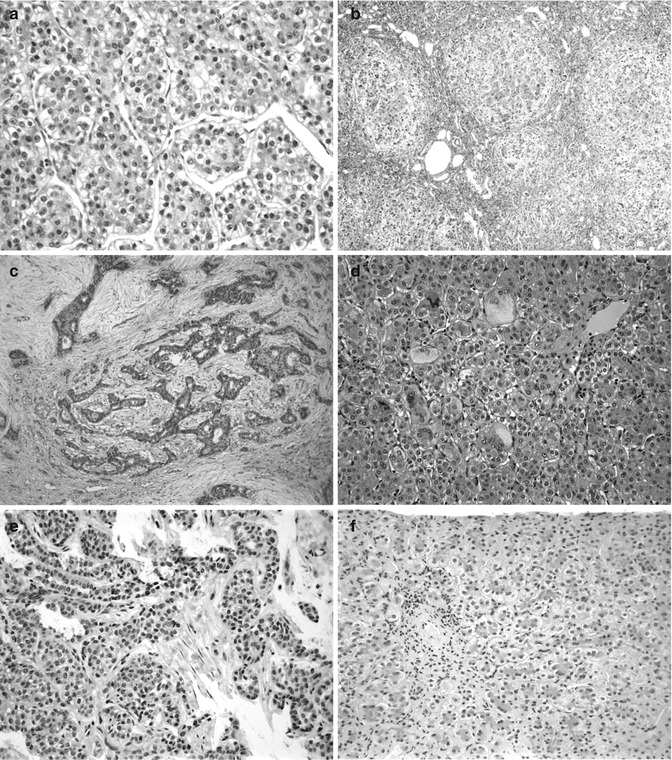
Fig. 14.4
Hepatic tumors arising in children with chronic cholestatic syndromes. (a) Hepatocellular carcinoma (HCC) in a 29-month-old boy has the typical trabecular growth pattern of a relatively well-differentiated HCC. (b) The liver elsewhere shows advanced biliary cirrhosis. The patient was demonstrated to have BSEP deficiency. (c) Cholangiocarcinoma is shown in a 4-year-old. The neoplastic ducts are found in close association with proliferating nonneoplastic bile ducts in this patient with biliary cirrhosis. (d) The patient presented at 2.5 months with neonatal hepatitis and marked giant-cell transformation. This patient had BSEP deficiency as confirmed by Strautnieks et al. (2008). (e) Cholangiocarcinoma is shown in an 8-year-old girl. (f) The patient presented at 5 months of age with paucity of intrahepatic bile ducts and cholestasis. The patient’s brother presented with the same neonatal hepatitis picture at the age of 8 months. The patients have Alagille syndrome
14.1.7 Metabolic Disease
The most frequent of the inborn errors of metabolism causing HCC in adults is hereditary hemochromatosis (Table 14.4). In children, type 1 hereditary tyrosinemia was the most common recessive condition, leading to liver cancer with HCC in as many as 18–37 % of affected children as early as 18 months (Roy and Finegold 2010). This is due to DNA damage from reactive oxygen species and accumulated mutagens (Erez et al. 2011) (Fig. 14.3f). Lindstedt discovered that blockade of an enzyme upstream in the tyrosine catabolic pathway with an organic herbicide (NTBC) could eliminate accumulation of mutagens (Holme and Lindstedt 1998). Since then, only those patients with subclinical hepatic injury and cirrhosis who fail to receive treatment in a timely manner progress to neoplasia (Holme and Lindstedt 2000). Other inborn errors of metabolism leading to hepatic neoplasia in children include alpha-1 antitrypsin deficiency (Hadzic et al. 2006), citrin deficiency, mitochondropathies, and various glycogenoses (see Chap. 13).
Table 14.4
Metabolic disease and HCC
Children |
Hereditary tyrosinemia, type 1 (FAH deficiency) |
Glycogen storage diseases, esp. type 1a (glucose-6-phosphatase deficiency) |
Chronic cholestasis syndromes: PFIC 2 (BSEP deficiency), Alagille syndrome (Jagged 1 deficiency), chronic TPN |
Mitochondrial – “Navajo” hepatopathy (MPV 17 mutation) |
Adults |
Hereditary hemochromatosis |
Alpha 1 antitrypsin deficiency |
Wilson’s (ATP7B mutation) |
Nonalcoholic steatohepatitis |
Glycogen storage disease (GSD) type 1a has led most often to adenomas, but on rare occasions, to focal nodular hyperplasia and HCC in adolescents and young adult males (Fig. 14.3c, d). Two siblings with GSD 1A, aged 12 years and 19 years, developed HB. Both patients had multiple tumors, at least one of which was an adenoma. Interestingly, neither patient had increased serum AFP. Recently, an adenoma and HCC were described in patients with brancher enzyme deficiency (GSD IV). Four adults with type III GSD or debrancher deficiency have been reported to have developed HCC (Roy and Finegold 2010), and an HCC has recently been observed in a 23-year-old woman with phosphorylase-B-kinase deficiency (type IX) (Manzia et al. 2011). A 5-year-old with Niemann–Pick, type C, developed HCC that was anaplastic and metastasized to the lungs (Birch et al. 2003).
14.2 Cytogenetics and Molecular Genetics
14.2.1 Cytogenetics (Table 14.5)
Table 14.5
Cytogenetic abnormalities in pediatric liver neoplasms
Hepatoblastoma (HB) | Trisomy 2, 8, 10 | Singly or in combination – 50 % | |
Trisomy 18 | Rare case reports | ||
Breakpoints | 1q12q21; 2q35-137 | With many deletions and translocations | |
(Translocation: der t(1;4)(q12;q34) | |||
HB and HCC | 1q excess by CGHa | 58–72 % | |
4q loses by CGH, or LOHb | 43–77 % | ||
Hepatocarcinoma | 8q excess by CGH or LOH | 48–77 % | |
16q LOH | 52–70 % | ||
17p loss by CGH or LOH | 49–51 % | ||
Fibrolamellar carcinoma | Aneuploidy | – | |
Hypertriploidy for 8, 20 | |||
Gain of 1q,8q, loss of 8q | |||
Hepatocellular adenoma | 6p+, 6q- | GSD1a | |
3 cases | |||
Mesenchymal hamartoma | Translocations of 19q13.4 | 7–8 cases | |
Aneuploidy | 2 of 8 cases | ||
Undifferentiated sarcoma (embryonal) | t(11;19)(q11;q13.4) | 1 patient with MH | |
Aneuploidy | Many, diverse | ||
Hemangioma, infantile hemangioendothelioma | del 6q(q21-927) | 1 case | |
Epithelioid hemangioendothelioma | t(1;3)(p36.3;q35) | 13 cases | |
Karyotypic abnormalities were found in 50 % of 111 HB from the Pediatric Oncology Group (Tomlinson et al. 2005). Forty-three cases had structural defects and 41 had numerical aberrations. Trisomy 20 and 2 were most common among the latter, followed by trisomy 8, singly or in combination. The most frequent abnormality involved 1q with breakpoints at q12–21, in 20 tumors. The corresponding translocations varied considerably but t(1;4) was found in seven cases, and the terminal region of 4q was most often involved, always in association with one of the common trisomies (Schneider et al. 1997). The four t(1;4) tumors were all in boys and at advanced stage (Fig. 14.5a). Translocation involving 4q32 is interesting because it occurs in 10 % of HCC and is an integration site for Hepatitis B virus. The 4q loss in 4 of 32 HB was found by CGH. The loss of heterozygosity (LOH) for loci on 1q and 1p has been shown in 7 of 32 HB each; LOH in both arms was found in three cases (Kraus et al. 1996). Three tumors had trisomies of chromosome 17, which may involve p53, HIC1, or neurofibromin loci. Consistent abnormalities in this region have not been observed by karyotyping. The only small-cell undifferentiated HB karyotyped had t(10;22), which was never seen in other HB, suggesting that these cells are not a precursor to fetal and embryonal cells in the tumors. Interestingly, we studied a sarcoma with t(22;22) (Fig. 14.24). 2q abnormalities are also observed in rhabdomyosarcoma.
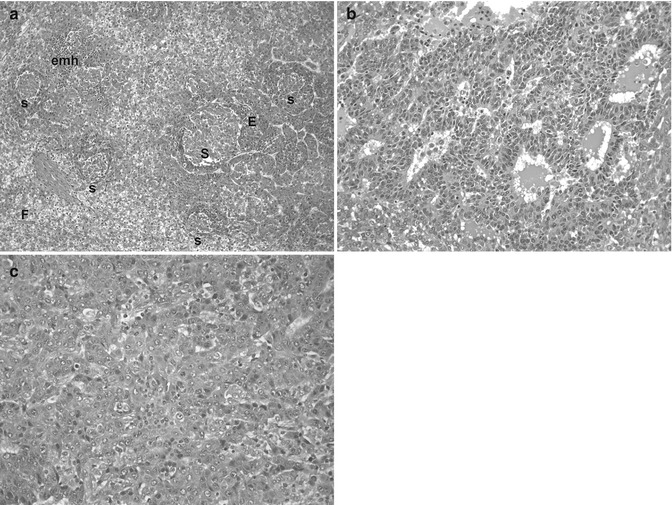
Fig. 14.5
Genetic alterations in hepatoblastoma. (a) This micrograph shows a typically mixed pattern containing mitotically active “crowded” fetal tissue (F), less well-differentiated embryonal components (E), and still less differentiated, small-cell undifferentiated portions (S). This tumor had a t(1;4) translocation in addition to +20 karyotype. There were no constitutional abnormalities. (b) This embryonal hepatoblastoma shows cells with a high nuclear/cytoplasmic ratio that form primitive tubules. The patient was an 11-month-old female with familial adenomatous polyposis whose HB was cured with chemotherapy and surgery. She required total colectomy at the age of 16 years. The tumor was 46XX with no 5q or other karyotypic abnormality. (c) This poorly differentiated epithelial hepatoblastoma in a 12-year-old patient with BRCA-2 mutation reveals highly pleomorphic cells that most closely resemble embryonal histology. Scattered normoblasts are present
In addition to these identifiable karyotypic abnormalities, many HB display double minutes. These fragments of chromosomes contain amplified DNA and are reflected in copy number variations found by single nucleotide pleomorphism (SNP) oligonucleotide arrays in most tumors (Fig. 14.6).
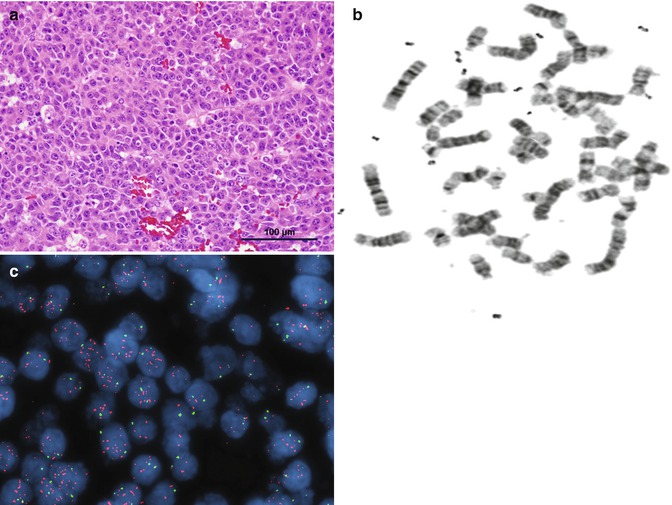
Fig. 14.6
(a) Embryonal HB. (b) Karyotype showing double minutes. (c) Fluorescence in situ hybridization (FISH) demonstrating marked amplification of the MDM4 gene on 1q32.1
14.2.2 Molecular Genetics (Table 14.6)
Table 14.6
Molecular genetics of hepatocellular neoplasms
Gene or locus | Abnormality | Tumor type |
|---|---|---|
FAP (APC) (5q 21.22) | Germ line mutations (FAP) or somatic mutations, LOH | HB, adenoma |
CDK 4 (12q 13–15) | ↑ Expression | HB |
Cyclin D1 (11q 13) | ↑ Expression | HB |
IGF2 (11 p 15.5) (BWS locus) | LOH or LOIa | HB |
RASSF1A (3p21.3) | Promoter methylation | HB |
Fibronectin, DLK1, TGFb1, MALAT1, and MIG6 | ↑ Expression | HB |
NOTCH 2 (1p13-p11) | ↑ Expression | HB |
GATA 4 (8p23.1-p22) | ↑ Expression | HB |
β-Catenin (3p 22-p21.3) | Mutations, ↑ expression | HB, adenoma, HCC |
Glypican 3 (GPC3) Xq26 | ↑ Expression | HB and HCC |
SERPINB3 (18q21.3) | ↑ Expression | HB and HCC |
HIC-1 (17p 13.3) | Hypermethylation, LOHb, 9 expression | HCC |
Telomerase (3q 26.3) | ↑ Expression | HCC |
P53 regions (17p 13) | Codon 249 mutation | HCC |
FAS (CD 95 L) | ↓ Expression | HCC |
E-cadherin (16q 22.1) | Hypermethylation ↓ Expression | HCC |
TGF α (2p13) | ↑ Expression | HCC |
Rb (13q14) | LOH | HCC |
Spondin2, PEG10, EDIL3, osteopontin | ↑ Expression | HCC |
TCF1 | HNF1α Inactivation Loss of LFABP1 | Adenoma |
14.2.2.1 Hepatoblastoma
The first suggestion that molecular genetic observations of HB may have prognostic significance was provided by Weber et al. (2000), who observed chromosomal gains of 8q and 20 by CGH more often in children who died of disease. The PLAG1 gene at 8q11.2–q13 is overexpressed in HB and contributes to IGF2 overexpression (Zatkova et al. 2004). CGH of a pre- and postchemotherapy HB showed 2q and 12 in a mixed tumor with fetal and embryonal epithelium that were not present in the large-cell anaplastic tumor that persisted after therapy and later metastasized. Chromosome 10 was enhanced in the recurrence. In two other tumors that had CGH before and after chemotherapy, only the posttreatment samples showed loss of 15q and gains of 18q and 22q. CGH results have been inconsistent, but gains of chromosome 1q, 2q, 8q, and 17 and losses of 4 are frequent. Since the first edition of this text, advances in molecular analysis have provided prognostic value as well as a better understanding of pathogenesis. For example, genome-wide SNP in GSD1A found simultaneous gain of 6p and loss of 6q in three of ten adenomas. Expression of IGF2R and LATS 1 candidate tumor suppressor genes on 6q was reduced over 50 % (Kishnani et al. 2009).
Children with the Beckwith–Wiedemann syndrome (BWS) have a relative risk of having an HB of 2280 (DeBaun and Tucker 1998), particularly in patients with hemihypertrophy. Eight percent of tumors in BWS are HB (Fig. 14.7a), and almost every other form of hepatic neoplasia has been observed in this syndrome (Fig. 14.23). The development of an embryonal malignancy in BWS patients is not as ominous as in other children. Vaughan et al. (1995) found 100 % disease-free survival in 13 affected children. Two of the tumors were advanced stage HBs that responded completely to chemotherapy. One patient with HB had an interstitial deletion of 11p, but 19 of 28 other BWS patients had no constitutional karyotypic abnormality. Of the nine cases with aberrations, six had triplication for 11p15. The p57KIP2 gene is located at 11p15.5 and is a negative regulator of cell proliferation. This is an imprinted gene with expression of only the maternal allele in normal cells. Mutation in the maternal allele has been observed in 4 of 49 BWS patients (Table 14.6). Many imprinted genes are dysregulated in HB, not only in 11p15; they include DLK1, PEG10, PEG3, BEX1, and RASGRF1. DLK1 is most heavily upregulated, especially in embryonal HB, reflecting its role in liver differentiation as a NOTCH ligand. Upregulation was not observed in a small undifferentiated HB (Lopez-Terrada et al. 2009), adding further evidence to the small cell not being a stem cell for HB (Badve et al. 2003).
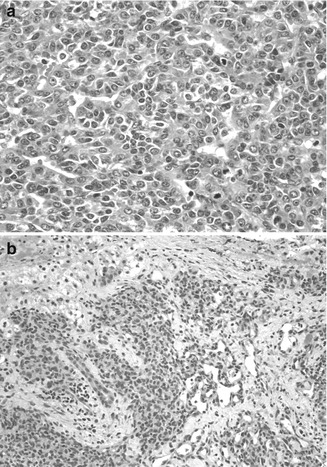
Fig. 14.7
Tumors in Beckwith–Wiedemann syndrome. (a) The embryonal hepatoblastoma is from an 8-month-old female. The embryonal cells are small and have a high nuclear/cytoplasmic ratio with ovoid nuclei and a tubular or rosette-like configuration. Mitotic activity is abundant. Rising serum AFP led to the discovery of a small tumor observed on a computerized tomographic scan. (b) A hemangioendothelioma was found in a 5-month-old patient with Beckwith–Wiedemann syndrome. The patient had masses in the liver and the lung, a large tongue, and a thymus gland that weighed 120 g (Specimen courtesy of Ken Barwick, MD. Wolfson Children’s Hospital, Jacksonville, FL)
Epigenetic modifications of the genome are of growing interest in tumors. Hypermethylation of DLK1–MEG3 is common in HB (Roy et al. 2010), and this could be particularly relevant to the striking excess of this embryonic tumor among survivors of prematurity, whose hepatocellular precursors are dividing rapidly while exposed to endogenous molecules that normally are eliminated by the placenta, as well as environmental challenges for which the immature liver is unprepared. Hypermethylation of both RASSFIA (Honda et al. 2008b) and MTIG (Sakamoto et al. 2010) has been found in HB with poor prognosis. Aberrant epigenetic silencing of SFRP1 by hypermethylation has been demonstrated in HCC, leading to Wnt activation and upregulation of c-Myc and cyclin D1 (Kaur et al. 2012).
Like Drut et al. (1992), we have observed hepatic hemangioendotheliomas in an infant with BWS (Fig. 14.7b). IGF2 is highly expressed during the proliferative phase of infantile hemangiomas and downregulated during regression, as shown with DNA microarray analysis, quantitative reverse transcription polymerase chain reaction (RT-PCR), and immunohistochemistry (Haas et al. 1986). Albrecht et al. (1994) found LOH for 11p15.5 in 6 of 18 HBs, one of which was from a BWS patient. This suggested loss of a tumor suppressor while preserving the growth stimulation activity of IGF2. Activation of the IGF axis and abnormal expression of molecules in this pathway are thought to be important in HB pathogenesis (Gray et al. 2000a) and have been proposed as HB therapeutic targets. IGF2 (insulin-like growth factor 2 or somatomedin A) is an imprinted gene located on chromosome 11p15.5 that regulates downstream phosphatidylinositol (PI) 3-kinase or mitogen-activated protein (MAP) kinase activation. Preferential induction of MAPK pathway signaling was reported in aggressive histologic epithelial HB subtypes (Adesina et al. 2009). Hartmann and colleagues (2009) identified strong p-AKT, p-GSK-3 beta, and p-mTOR expression in most HB tumors, as well as a PIK3CA gene mutation in one HB. Downregulation of mRNA for H19, a noncoding candidate for the suppressor role, was found in 10 of 12 HB by Hartmann et al. (2000). The same authors noted upregulation of p57 in 9 of 12 tumors, suggesting reactivation of the paternal allele in those tumors. Interestingly, LOH for 11p15 markers in an HB was found in a child with FAP who had lost the maternal APC gene in the tumor while retaining a mutant allele from the father. This work refines the first observation of LOH of 11p in diverse tumors of BWS patients by Koufos et al. (1985). There have been many studies of gene expression in the 11p15 region (Table 14.6). The tetraspanin gene CD81 that complexes with integrins and thereby influences cell–cell interactions and LSP1, which binds f-actin and regulates intracellular calcium, are also found at this locus. Insulin and IGF2 are stimulants of embryonal cell growth, so their upregulation or loss of imprinting in HB is suggestive of an early genetic contribution to fetal oncogenesis (Honda et al. 2008a). Elevated serum levels of hepatocyte growth factor (HGF) have been observed in HB patients at the time of diagnosis and proposed to be at least partly responsible for tumor progression. HGF/c-Met-related activation of beta-catenin was reported in a proportion of wild-type CTNNB1 tumors (Purcell et al. 2011). Hence, HGF and its receptor c-Met are candidates for targeted therapy in these tumors (Fig. 14.8).
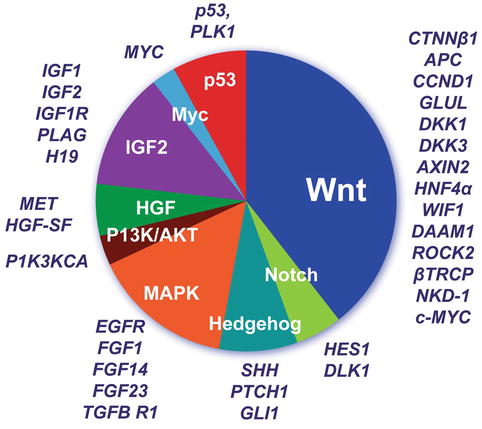
Fig. 14.8
Hepatic differentiation is governed by the canonical WNT and NOTCH pathways. Gene expression correlates with particular beta-catenin mutations and histopathology of HB (Lopez-Terrada et al. 2009)
HB occurs in children of families with heritable polyposis (FAP) at a frequency of 1,000–2,000 times that of the population at large (Fig. 14.5b). De Chadarevian et al. (2002) found a constitutional inversion of chromosome 5 in a family with FAP and two siblings with hepatocellular neoplasia. Nevertheless, no karyotypic alterations in tumors have been detected at 5q21. Oda et al. (1996) found mutations in APC in 8 of 13 sporadic HB, with no abnormality in the nontumoral liver. The kinds of germ line mutations of APC in patients with HB are not different from families without liver tumors; all the survivors of HB have later developed colonic polyposis. Other hepatobiliary tumors have been reported in FAP cases, including HCC in a 9-year-old boy and fibrolamellar carcinoma in a 15-year-old girl, adenoma in a 2-year-old child with biallelic inactivation of the APC gene in the tumor, bile duct adenoma, and gallbladder adenomatous proliferation and dysplasia. We have elsewhere depicted the simultaneous presence of adenoma and HB in an 11-year-old (Hadzic and Finegold 2011).
The APC protein regulates beta-catenin, an E-cadherin-binding protein normally involved in intercellular adhesion. When APC is mutated, beta-catenin accumulates in cells, where it stimulates expression of Wnt pathway. Wnt is important for the embryologic differentiation and zonation of the liver (Thompson and Monga 2007). Activating mutations in beta-catenin have been observed in 50–90 % of HB (Koch et al. 1999) and in 20–26 % of HCC (Lopez-Terrada et al. 2009). In the 30 % of HB found not to have chromosomal gains or losses by CGH by Buendia, all had beta-catenin mutations (Nhieu et al. 1999). Mutations of beta-catenin lead to nuclear translocation, activation of diverse genes, and increased cell proliferation. With immunohistochemistry, a shift of beta-catenin from its normal localization at the hepatocyte plasma membrane to the nucleus is found, especially at the invasion fronts, whereas well-differentiated fetal cells often preserve membranous staining (Table 14.7).
Table 14.7
Immunohistochemistry for liver tumors
Primary liver tumors | |||
Antibody | Target | Issues | |
Hepatoblastoma (epithelial) and HCC | αFP | Immature-maturing hepatoblast | Some even WD tumors are negative |
Hep Par | Differentiated hepatocyte | Normal HC + | |
Beta–catenin | Nuclei, diffuse cytoplasm = neoplasia | Benign HC tumors +; very useful post-chemotherapy | |
Glypican 3 | Diffuse cytoplasm = neoplasia | Very useful after chemo | |
Epithelial cytoplasm | Reactive HC, FNH maybe + | ||
Cholangioblastic HB and cholangiocarcinoma | CK19, CK7 | Biliary epithelium | |
CE | |||
CA19-9 | |||
Stem cell (HCC) | CD133, EpCAM, DLK1, DKK1 | Precursor cells | – |
Fibrolamellar HCC | Nestin, HNF1α, CK19, CD44 | ||
Mixed HB | Vimentin, EMA | Osteoid elements | – |
Teratoid HB | GFAP, HMB45, Beta–HCG | Polyphenotypic differentiation | – |
Teratoma | |||
Small-cell undifferentiated and rhabdoid tumor | INI | Negative nuclei | Very rarely + |
Cytokeratin + vimentin | Dual cytoplasmic positivity | Desmin will be negative | |
Endocrine carcinoma | Chromogranin, synaptophysin | – | – |
Nested stromal–epithelial tumor | ACTH, WT1, SMA, NSE, CD56, vimentin, EMA, CK | Duct relationship, stromal and secretory cells | Epithelial-mesenchymal transition |
Adenoma | (a) L–FABP1 | (a) TCF1 mutation | (c) Pre–HCC |
(b) CRP, SAA | (b) Inflammatory | ||
(c) Beta–catenin, glutamine synthetase | (c) WNT pathway | ||
Rhabdomyosarcoma | Desmin, myogenin, MyoD1 | – | – |
Inflammatory myofibroblastic tumor | AlK1 | – | – |
CD138 | |||
Infantile hemangioma (hemangioendothelioma) | Glut1 | – | RBC are + |
CD31, CD34 | |||
Epithelioid HE | CD31, SMA, VWF | – | – |
Angiosarcoma | CD31, CD34, VWF | – | Glut1 negative |
Angiolipoma – PEComas | HMB45, TFE3, cathepsin K, SMA | – | – |
Undifferentiated (embryonal) sarcoma | Vimentin, α1AT, CK | – | Glypican 3-residual epithelium |
Variable myogenin, S-100, SMA | |||
Metastatic or systemic tumors | |||
Leukemia | CD45, LCA, Platelet GP | – | – |
Megakaryoblastic | |||
Intra-abdominal desmoplastic small-cell tumor | Desmin, EMA | – | – |
CK | |||
NUT midline carcinoma | CD52 | – | Useful for unknown primary with metastasis to liver |
Lopez-Terrada et al. (2009) investigated the canonical Wnt signaling pathway in HBs and correlated the histologic subtype, beta-catenin immunostaining, and mutation status of the CTNNB1 gene (Fig. 14.8). All tumors with embryonal or small-cell histology showed either point mutations in exon 3, deletions within or confined to exon 3, or showed no CTNNB1 mutations. By contrast, HBs with pure fetal epithelial histology demonstrated predominantly large deletions of CTNNB1 including the entire exon 3 and most of exon 4. Immunohistochemistry demonstrated a correlation between the presence of CTNNB1 mutations confined to exon 3 and increased nuclear translocation of beta-catenin, particularly in the higher-grade tumor components, whereas HBs with large CTNNB1 deletions showed nuclear translocation of beta-catenin confined to a much smaller proportion of tumor cells or only the normal staining of cell plasma membranes. Ueda and colleagues recently reported Wnt activation in a small subset of HBs with wild-type beta-catenin, associated with high expression levels of telomerase reverse transcriptase (TERT), chemoresistance, and poor prognosis.
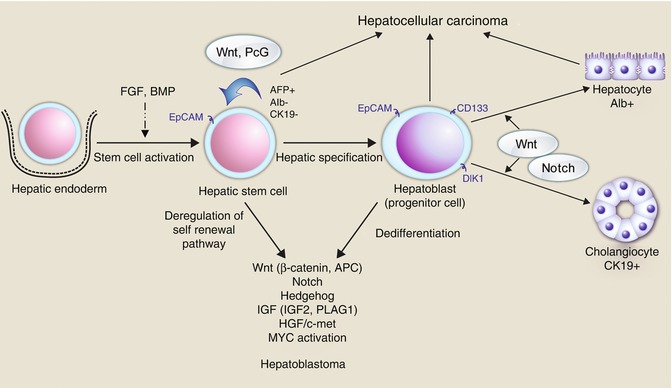
Fig. 14.9
The many pathways and genes involved in hepatic embryogenesis and maturation and the relative contributions to neoplasia (Chavan et al. 2012)
The NOTCH developmental pathway is involved in stem cell self-renewal and is necessary for cholangiocytic lineage differentiation. NOTCH pathway is also activated in some cancers, including HB. DLK1 (delta-like protein/preadipocyte factor 1/fetal antigen 1 or DLK/Pref1) is a NOTCH ligand which is highly expressed in bipotential transit amplifying oval cells and upregulated in hepatocellular tumors, including HCC and HB. NOTCH2 overexpression has been recently reported in HB (Litten et al. 2011).
Another pathway in development and liver regeneration is Sonic Hedgehog, which is activated in HCC (Huang et al. 2006). Increased transcription of GLII and PTCH1, HHIP gene promoter methylation, and a strong inhibitory effect on proliferation by blocking Hh signaling have been reported using HB cell lines. Increased expression of Sonic Hedgehog (SHH), Patched (PTCH), and the transcription factor GLI has also been detected by immunohistochemical analysis in HB specimens (Eichenmuller et al. 2009). However, no PTCH1 mutations were identified in a recently reported series of HBs (Chavan et al. 2012).
HBs derive from pluripotent stem cells (hepatoblasts), defined by their ability to differentiate into multiple cell lineages, self renew, and regenerate. Tumor heterogeneity characteristic of HB may be explained either by ongoing mutagenesis or by their origin from a stem cell arrested at different developmental stages and able to give rise to diverse forms of cancer, or by a combination of both (Fig. 14.9).
Expression profiling analysis has also been reported in HB by several groups. Nagata and colleagues (2003) identified 26 differentially expressed genes in HBs, when compared to the adjacent liver tissue, including 4 located on the 1q21 chromosomal region (ETV3, TPR, CD34, and NR113), and other genes encoding cell division and growth regulators (IGF2 and IGFBP4). Gene expression study of HB and the adjacent liver identified 6 genes (IGF2, Fibronectin, DLK1, TGFb1, MALAT1, and MIG6) differentially overexpressed in HB (Luo et al. 2006).
Comprehensive HB profiling studies by Cairo and colleagues (2008) described liver stem-like phenotype resulting from a combined Wnt/beta-catenin and c-Myc signaling, recapitulating a developmental stage with a profile similar to a hepatic stem/progenitor. Cairo et al. (2008) developed a 16-gene signature whose altered expression correlated with great significance to the prognosis of HB. The most dramatically expressed gene was DLK1 at 61.7× that of the normal liver, a member of the Wnt pathway, whereas ANNEXIN 10 was most significantly downregulated (tenfold). Figure 14.8 depicts the many genes and pathways in which abnormal expression has been reported.
miR profiling of HBs and HCCs demonstrated that Myc-driven reprogramming of miR expression contributes to an aggressive hepatic progenitor cell tumor phenotype and revealed a four-miR signature that may be useful to stratify HB patients (Cairo et al. 2010) (Table 14.8).
Table 14.8
MicroRNA and liver cancer
Up (4) and down (1) expression of miRNAs in HB versus host liver (Magrelli et al. 2009) |
Dicer1 knockout mice have increased hepatocellular growth and apoptosis, with spontaneous HCC by 1 year (Dicer1 is required for microRNA processing) (Sekine et al. 2009) |
miR-122 is abundant in primary HC but not HCC cells. It targets oncogenes and mediates actions of Sorafenib versus HCC (Bai et al. 2009) |
Expression of cyclins in HB is linked to the Wnt pathway. Significant downregulation of cyclin D1 was detected in four of seven tumors, whereas cyclin D2 and D3 were overexpressed versus nonneoplastic tissue using quantitative RT-PCR for mRNA and Western blotting for proteins (Iolascon et al. 1998). With immunohistochemistry, cyclin D1 was overexpressed in comparison to adjacent nontumoral liver in 13 of 17 cases, and cdk4 was increased in 15–17. In 11 patients both were overexpressed. Expression of the cdk inhibitor p27 has been lost in many aggressive neoplasms. We found a similar result in 27 HB (Brotto and Finegold 2002). The most interesting correlation was seen in the well-differentiated fetal component that has been associated with a favorable outcome when present as a pure population but not necessarily when admixed with other cell types (Haas et al. 1989). Fetal-type tumor cells that remained after chemotherapy in ten cases showed an average decline in the percentage of p27-positive nuclei from 75 to 12 % (Fig. 14.10a–d). All ten cases had decreased expression, and it was completely absent in two. Embryonal epithelium before and after therapy had 23 % positive cells. Small, undifferentiated cells in 3 cases were p27 negative in 2 and only 5 % were positive in the other. Stem cell markers were identified histochemically among fetal hepatoblasts rather than small cells by Fiegel et al. (2004).
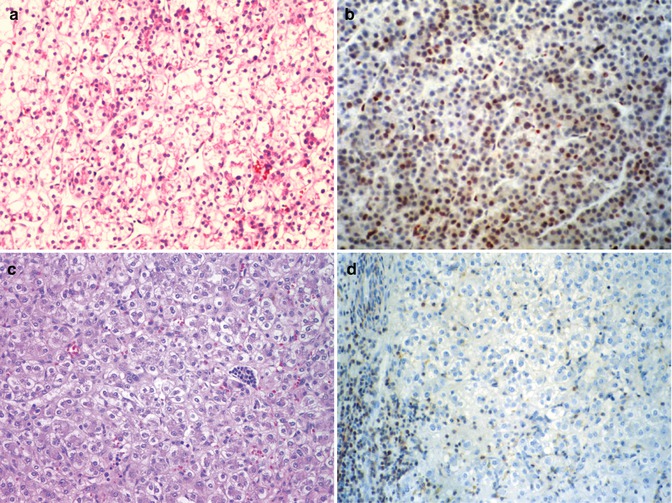
Fig. 14.10
P27 in fetal hepatoblastoma. (a) The biopsy specimen of a stage III hepatoblastoma shows well-differentiated fetal tumor cells with a low nuclear/cytoplasmic ratio and glycogen-rich cytoplasm. (b) The P27 immunostain is positive in the nuclei of approximately 60 % of the fetal hepatoblasts. (c) Following an excellent response to chemotherapy, the patient’s tumor became resectable, but there was residual fetal hepatoblastoma. (d) The tumor cells fail to stain for P27. The positive cells on the left represent circulating leukocytes
The first application of CHIP technology to screen for mRNA expression in HB revealed a surprise, which was BRCA-2 expression in three cases (Schofield et al. 1999) (Fig. 14.5c). BRCA-2 is constitutionally mutated in familial breast and ovarian cancers and was mutated in 3 of 60 HCCs. Interestingly no APC expression was found with CHIP probes in the three tumors. BRCA-2 is a member of the Fanconi gene family and involved in breakpoint repair. Antibody to BRCA-2 stained six of six HBs but not fetal liver.
14.2.2.2 HCC
Chromosome 1q, 4q, 8p, 8q, 16p, and 17p abnormalities have been most commonly identified in HCC, relying mainly on CGH and microsatellite polymorphisms (Table 14.5). Few of the many studies include pediatric cases, most of which are not associated with chronic infection and cirrhosis, as in adults. From 58 to 72 % of HCC had increased copies of 1q. The high incidence of 8q excess may account for increased c-Myc expression, as it is located at 8q24 (Wong et al. 1999). Fluorescence in situ hybridization (FISH) showed that polysomy for chromosome 1 was the most common abnormality in HCC and trisomy 8 was next (Nasarek et al. 1995). The most frequent abnormalities found with CGH were of 4q, with decreased copies in 43–70 % and LOH in 77 % of cases. 6q, where many HCC have LOH or decreased copies, is not a site of abnormality in HB. Interestingly, 20, 8, and i(8 q10), as well as rearrangement of 1, were found in the cytogenetic analysis of a metastatic fibrolamellar carcinoma, resembling HB (Lowichik et al. 1996). CGH has been helpful in distinguishing well-differentiated HCC from adenomas, which do not show any of the several abnormalities in HCC (Wilkens et al. 2001). LOH for a DNA repair gene has led to microsatellite instability in HCC.
The rapidly expanding application of molecular genetic analyses of HCC has provided much data relevant to pathogenesis and prognosis. Table 14.6 shows the genes that have received the most attention. For most genes the proportion of carcinomas in which abnormalities of expression have been demonstrated is generally less than 50 %, and many of the defects occur only in the advanced stage of the disease, when many secondary mutations have accumulated. As in a growing number of cancers, defects in DNA repair contribute to HCC as in Fanconi anemia, BRCA-2 mutation, and Bloom syndrome (Jain et al. 2010).
Molecular profiling of HCC has resulted in the recognition of stem-cell signatures and characteristic profiles associated with response to therapy and survival. Thorgeirsson and colleagues demonstrated that HCCs with the cancer stem-cell (CSC) profile had worse prognosis, similar to what had been reported in other tumor types (Lee et al. 2006). A summary of work from many laboratories by Andrisani et al. (2011) provides the following expression data that strengthens the correlation with poor prognosis in HCC: increased expression of EpCAM, CK19, DLK1, DKK1, PROM1 (CD133), AFP, and Myc and decreased expression of UGT2B7 and CYP3A4. Just as in HB, microinhibitory RNAs (miRNA) have become increasingly relevant to HCC (Cairo et al. 2010) (Table 14.8). Detection of miR-15b and miR-130b in the serum of patients with HCC by RT-PCR was strikingly reduced after resection, particularly in patients with normal serum AFP (Liu et al. 2012).
The most consistent alteration in HCC has been codon 249 mutations of p53 (TP53 gene), which was found in 67 % of patients exposed to aflatoxin in Mexico, Mozambique, South China, and Africa. The frequency of p53 mutations elsewhere in the world is lower and involves other loci in the gene. LOH for p53 was found in 57 % of HCCs and 63 % of adjacent cirrhotic nodules, indicating premalignant loss of the tumor suppressor in some cases (Kishimoto et al. 1997). The importance of p53 is to regulate cell division via p21 WAF by sensing abnormalities in the DNA and preventing defective cells from dividing. The overexpression of p53 in HB and other liver malignancies, when detected immunohistochemically, does not signify mutations in the gene (Kennedy et al. 1994). p53 also activates expression of HIC-1, a recently discovered candidate for tumor suppression in a nearby chromosomal region (17p 13.3) (Wales et al. 1995). HIC-1 mRNA is markedly reduced in precancerous liver nodules of chronic hepatitis and cirrhosis and even more so in HCC, especially in poorly differentiated tumors. This is due to hypermethylation at the D17S5 locus on 17p. There was LOH at the locus in 54 % of carcinomas. Reduced expression of E-cadherin in HCCs also was attributable to methylation of the promoter on 16q 22.1 (Kanai et al. 1997). High levels of CpG methylation also account for loss of the tumor suppressor p16INK4 expression in half of the HCC (Hui et al. 1996). Silencing of the JAK/STAT regulator, SOCS-1, with aberrant methylation was found in 65 % of primary HCC (Yoshikawa et al. 2001). The Mdm2 proto-oncogene was overexpressed in 6 of 23 HCC. Mdm2 is known to downregulate p53 activity by competing with it, but it appears to have a p53-independent role in carcinogenesis because transgenic mice deficient in p53 also develop many sarcomas. No amplification of Mdm2 was found in 19 HB (Ohnishi et al. 1996). However Mdm4, whose product interacts with Mdm2 to inactivate p53, is overexpressed in 35 % of HB, especially in embryonal cells (Fig. 14.6c and Roy et al. 2012). The tumor suppressor, P21, is similarly inactivated (Hui et al. 1996) by excess Mdm4. When p53 mutations in other loci were detected in a group of HCC that were treated with surgical excision, the mean patient survival was reduced from 60 months to 15 months, compared to patients with wild-type p53 in their tumors (Honda et al. 1998). The presence of antibody to p53 in the serum of patients with HCC (32 % of 86 patients) was predictive of shorter survival (Shiota et al. 1997).
Independent clonal origin of multiple HCC in the same liver has been demonstrated with different patterns of LOH in the separate tumors (Tsuda et al. 1992). This observation was superbly elaborated by Gerlinger et al. (2012) in relation to the marked intratumoral heterogeneity for mutations in several tumor suppressors. As a result of the divergencies, expression signatures from one part of a tumor may suggest a poor prognosis while the opposite may be concluded from a second sample. This careful study emphasizes the potential danger of “personalized” cancer gene therapy from a single small sample (Fig. 14.11). When LOH for a particular locus is observed in noncirrhotic livers, it may have more significance. That is the case for 5q LOH in five of seven patients from England (Gray et al. 2000b). Nine cirrhotic patients with HCC failed to show this abnormality, but both groups had LOH at 17p13 (the p53 and HIC 1 loci). The 5q locus was at q35-qter, distant from the APC and MCC genes at 5q 21–22.
Fig. 14.11
Size of biopsies compared to a typical HB
Telomerase activity was not detectable in a normal liver but was increased in 17 % of nontumoral tissue from patients with HCC and was present in 38 % of the tumors. It was more prevalent in moderately differentiated than well-differentiated tumors. Eighty-six percent of large regenerative nodules in patients without carcinoma had telomerase activities equal to carcinomas (Hytiroglou et al. 1998). Telomerase activity in the nontumoral tissues was associated with early recurrence (Ohta et al. 1997).
The Hippo pathway of growth control in Drosophila has received much attention recently in diverse human cancers (Liu et al. 2010). Even without amplification of the gene on 11q22, overexpression of nuclear YAP (Yes-associated protein) is observed in many HCC (Li et al. 2012) along with its targets, glypican 3, CTGF, and survivin. YAP1 phosphorylation is downregulated in 30 % of HCC, leading to a loss of targeted Mst1, a tumor suppressor (Zhou et al. 2009). This pathway also influences contact inhibition and hepatocellular growth through E-cadherin (Kim et al. 2011). And this provides for a cytoplasmic interaction with beta-catenin and the Wnt pathway. YAP also associates with beta-catenin in the nucleus to activate gene expression and excessive hepatocellular replication (Imajo et al. 2012). Thus, each new observation of genes involved in the control of the cell cycle, apoptosis, and cell–cell interaction points to nodes of interaction among the pathways and the likelihood of redundancy as well as additive effects.
14.2.2.3 Hepatocellular Adenoma
The development of benign as well as malignant hepatocellular proliferation in patients with constitutional defects in DNA replication and repair (Fanconi anemia), the Wnt pathway (familial polyposis), or metabolic disease (GSD1a) has been illustrated (Fig. 14.3). Adenomas characterized by Wnt pathway disturbances, including beta-catenin activation, were particularly prone to the simultaneous or subsequent presence of HCC in the large series from France (Bioulac-Sage et al. 2009). This group delineated a second set of adenomas due to mutations of TCF1 that inactivate HNF1α and could be identified immunohistochemically and by RT-PCR to have lost FABP1 expression. A third form of adenoma was characterized by steatosis and inflammation (Fig. 14.12c), especially in obese and alcoholic adults and often hemorrhagic and telangiectatic. Immunohistochemistry revealed serum amyloid A only in this group. Restoration of G6Pase activity by adenovirus-mediated gene therapy to the liver in mice lacking the gene not only corrected their hypoglycemia and hepatic steatosis but prevented adenoma formation, providing an exciting prospect for humans with this condition (Lee et al. 2012).
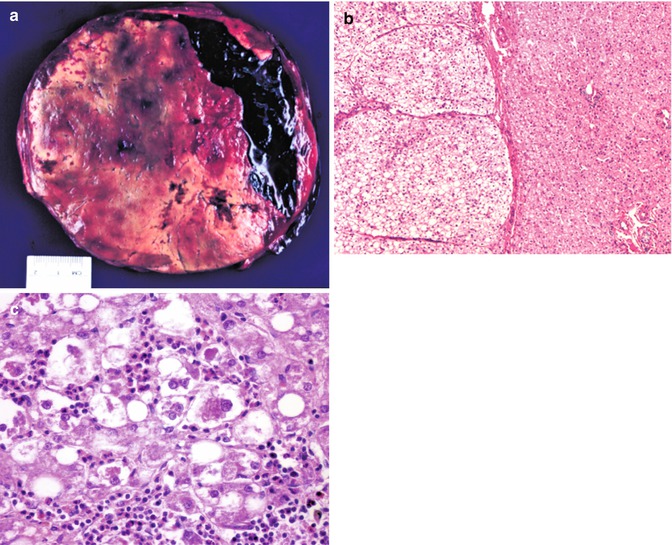
Fig. 14.12
Hepatocellular adenoma. (a) This resection of a well-circumscribed mass is from a 15-year-old female who presented with severe abdominal pain and sudden development of anemia. The resection shows a major hemorrhage into the benign tumor. This patient had been taking oral contraceptives. (b) The border between adenoma on the left and the normal liver on the right is well demarcated in this instance. A thin capsule is present. No portal tracts and accompanying bile ducts are found within the adenoma. The cells, rich in lipid, are slightly larger than those of the normal liver. This patient has glycogen storage disease type Ia. (c) Higher magnification view of another patient’s adenoma reveals foci resembling alcoholic hepatitis. Mallory material is present in the cytoplasm of some hepatocytes to which there is a neutrophilic reaction
14.2.2.4 Mesenchymal Tumors
Chromosomal aberrations in hepatic neoplasms of mesenchymal origin are shown in Table 14.5. It is particularly interesting that in contrast to HB, in which triploidy for chromosomes 20 and 2 are the most common findings, those chromosomes are frequently lost in the undifferentiated or embryonal sarcoma (Lauwers et al. 1997). It is also noteworthy that mesenchymal hamartomas, which some authors attributed to a vascular accident in utero, have a characteristic translocation involving t(19q 13.4), consistent with a neoplastic process rather than a post-injury reparative phenomenon (Shehata et al. 2011). This is clinically important because some publications have advocated conservative management, such as marsupialization of cysts without resection. Failure of marsupialization does not necessarily lead to sarcoma but to recurrence. At least seven cases of mesenchymal hamartoma have been reported to evolve into a sarcoma, and the translocation t(11;19)(11q,13.3–13.4) was demonstrated in one sarcoma (Rajaram et al. 2007). An association between mesenchymal hamartoma and placental mesenchymal dysplasia has been reported. Androgenic biparental mosaicism, as observed in the latter, has now been demonstrated in the liver lesion (Lin et al. 2011). This is particularly interesting because both conditions occur in the Beckwith–Wiedemann syndrome, which often involves uniparental disomy of the imprinted locus on 11p15. We have described a BWS patent with MH in infancy whose embryonal HB was overlooked and 4 years later was found to have HCC and cholangiocarcinoma in the hepatectomy specimen (Hadzic et al. 2011) (Fig. 14.23f).
A variety of molecular genetic alterations in hemangiomata associated with particular syndromes, such as von Hippel–Lindau and cerebral cavernous angiomas (KRITI/CCM1 mutation), have yet to elucidate the pathogenesis of hepatic hemangioendotheliomas. Whether the observation of VEGF receptor mutations in 2 of 15 hemangioendotheliomas of children will be applicable to liver lesions remains to be demonstrated (Walter et al. 2002). Hepatic hemangiomas were part of the maldevelopment complex in mice heterozygous for the tuberous sclerosis gene TSC 1. The tumors lost the normal allele. Hemangiomas occur in infants with BWS (Fig. 14.7b) independently or associated with mesenchymal hamartoma (Hadzic et al. 2011). One of three epithelioid hemangioendotheliomas found to have a t(1;3)(p36.3;q25) translocation with WWTR1–CAMTA1 fusion arose in the liver (Errani et al. 2011). Angiosarcomas induced by thorotrast radiation display K-ras mutations.
14.2.2.5 Inflammatory Myofibroblastic Tumor
The genetic abnormalities of inflammatory myofibroblastic tumor include clonality by karyotyping, 9p deletion, and 2p23 abnormalities with anaplastic lymphoma kinase (ALK) rearrangements (Coffin et al. 2001). The latter occurred in slightly younger children and recurred more often. Malignant transformation was observed twice in children with ALK1 abnormalities and twice with aneuploidy but no ALK abnormality. A more aggressive variant of IMT arising in the abdomen has been designated as epithelioid inflammatory myofibroblastic sarcoma and is characterized by ALK expression on or near the nuclear membrane (Marino-Enriquez et al. 2011). Nine of the eleven patients had ALK rearrangements by FISH and three had RANBP2–ALK fusion by RT-PCR.
14.3 Clinical Manifestations
The presenting symptom of virtually all liver tumors in children is abdominal swelling secondary to hepatomegaly. The enormous functional reserve of the liver, its location, and size permit long periods of occult growth, so that many tumors are very large when first detected and may have disseminated. Abdominal pain is the next most common, followed by gastrointestinal symptoms, including vomiting and anorexia. Failure to gain weight or loss may be noticed first. Rare patients may present with fractures due to osteoporosis or virilization due to gonadotropin secretion, these being associated with HB and less often with HCC. Jaundice is infrequent and limited to masses compressing or arising from the major bile ducts. Botryoid rhabdomyosarcoma (Fig. 14.13) and inflammatory myofibroblastic tumor (Fig. 14.14) are most often responsible (Kim et al. 1996). Pruritus may herald jaundice. Rupture of large or highly vascular HB and adenomas may lead to exsanguination. Although HB can have extensive telangiectases (Fig. 14.19c), they do not contribute to signs of high-output heart failure, as observed in 15 % of infants with hemangioendotheliomas, and rarely with mesenchymal hamartomas.
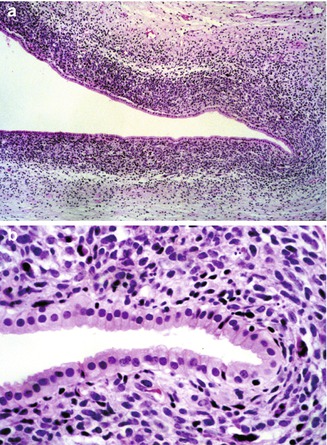
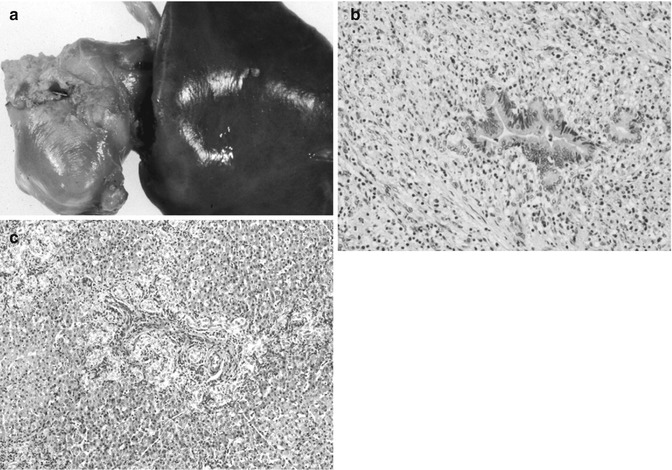
Fig. 14.13
Rhabdomyosarcoma of liver. (a) A 7-month-old male infant presented with signs of obstructive jaundice. A biopsy specimen revealed small undifferentiated cells forming a cambium layer surrounding major bile ducts near the hepatic hilum. (b) Higher magnification reveals little evidence of myogenic differentiation in routine sections, but immunostains were positive for myogenin
Fig. 14.14
Inflammatory myofibroblastic tumor. (a) The patient was a 4-month-old who presented with evidence of obstructive jaundice. Imaging studies revealed a mass at the hilum of the liver and fine-needle aspirates showed only unremarkable hepatocytes, the extreme sclerosis of the mass having diverted the needle into the surrounding hepatic parenchyma. (b) Hepatic portoenterostomy was performed successfully. This section shows a hepatic duct branch completely surrounded by a mixture of inflammatory cells and proliferating myofibroblasts. (c) The liver elsewhere was biopsied and shows a pattern typical of obstructive jaundice with extensive bile duct proliferation. The patient’s bile flow resumed within 24 h of surgery, and he has had no return of jaundice in 3 years. However, some portal hypertension is evident
When confronted with these symptoms, it is useful to consider the age at which liver tumors tend to occur (Table 14.9). Exceptions are frequent, but age can serve as a guide when the presenting symptoms lack specificity. Sixty-six percent of HBs in the POG–COG series from 1986 to 2011 were manifested by the second year and 11 % before 6 months of age (Table 14.10). Approximately 50 % of those in infants were congenital, given their size when discovered by 2–3 months. Isaacs’ review (2007) of liver tumors diagnosed prenatally (56) or in newborns (138) from 1970 to 2005 found 117 hemangiomas (60 %), 45 mesenchymal hamartomas (23 %), and 32 HBs (16.5 %); 5.6% of HBs occurred after age 5 years. HCC have been observed as early as 6 months. Seven examples of mixed HB and HCC have been observed in our series at a mean age of 8.5 years. These may be the equivalent of the so-called “transitional” or highly aggressive HBs of the older child and adolescent reported by Prokurat et al. (2002). Perinatally acquired HBV was responsible in three instances. Yolk sac tumors are embryonal in character, but there are several case reports of yolk sac tumors in older adults. They are rarely present as part of an HB. Systemic malignancies and metastatic disease must be considered at all ages, as hepatomegaly due to megakaryoblastic leukemia, Langerhans cell histiocytosis, and neuroblastoma are important sources of confusion with HB in infancy and intra-abdominal small-cell desmoplastic tumors later in childhood (Fig. 14.15).
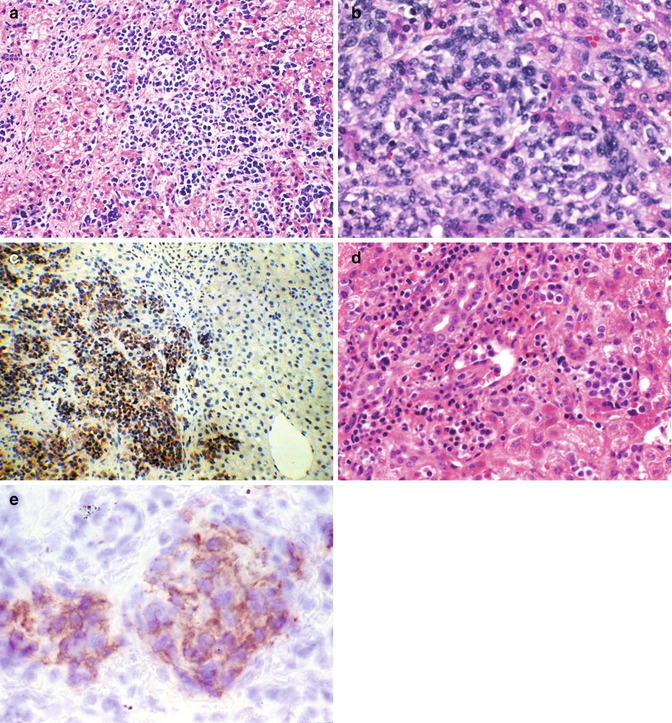
Table 14.9
Usual age of presentation of liver tumors in children
Age | Benign | Malignant |
|---|---|---|
Infancy (0–1 years) | Hemangio(endothelio)ma AV malformation Mesenchymal hamartoma Teratoma | Hepatoblastoma, especially small-cell undifferentiated Rhabdoid tumor Yolk sac tumor Langerhans cell histiocytosis Megakaryoblastic leukemia Disseminated neuroblastoma |
Early childhood (1–3 years) | Hemangioendothelioma Mesenchymal hamartoma Inflammatory myofibroblastic tumor | Hepatoblastoma Rhabdomyosarcoma |
Later childhood (3–10 years) | Adenoma; Focal nodular hyperplasia Angiomyolipoma | HCC Embryonal sarcoma (undifferentiated) Angiosarcoma |
Adolescence (10–16 years) | Adenoma; FNH, nodular regenerative hyperplasia Biliary cystadenoma | Fibrolamellar/HCC Nested stromal–epithelial tumor Hodgkin’s disease/lymphoma Leiomyosarcoma |
Table 14.10
Children’s oncology group hepatoblastomas in 25 years (1986–2011)
Stage | Age | Gender | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Total | I | II | III | IV | 0–1 years | 1–2 | 2–5 | >5 | M | F | |
Epithelial HB | 450 (62.5 %) | 103 | 22 | 224 | 77 | 130 | 159 | 140 | 21 | 283 | 158 |
Pure fetal | 74 (10.4 %) | 21 a | 1 | 33 | 9 | 25 | 14 | 16 | 8 | 49 | 25 |
Pure embryonal | 11 (1.5 %) | 2 | – | 7 | 2 | 4 | 1 | 5 | 1 | 7 | 4 |
Mixed | 127 (17.6 %) | 34 | 6 | 63 | 17 | 51 | 46 | 21 | 5 | 64 | 63 |
Teratoid | 39 (5.4 %) | 6 | 3 | 22 | 8 | 3 | 17 | 16 | – | 17 | 14 |
Pure small cell | 7 b (0.1 %) | 1 | – | 5 | 1 | 6 | – | 1 | – | 6 | 1 |
Rhabdoid | 6 (0.1 %) | – | – | 1 | 3 | 6 | – | – | – | 5 | 1 |
HB/HCC | 5 (0.1 %) | 1 | – | 3 | 1 | – | 1 | – | 4 | 3 | 1 |
Total | 719 c | 168 (25 %) | 32 (4.7 %) | 358 (53 %) | 118 (17 %) | 225 (32 %) | 237 (34 %) | 199 (28 %) | 39 (5.6 %) | 417 (61 %) | 267 (39 %) |
Fig. 14.15
Childhood malignancies that may be confused with hepatoblastoma. (a) Neuroblastoma is seen in the liver of a 7-month-old infant. Small blue, round cell tumors involving the liver include embryonal rhabdomyosarcoma, the blastematous form of Wilms’ tumor, lymphomas, and the small-cell undifferentiated version of hepatoblastoma. Immunochemistry is a simple and rapid way to distinguish among these possibilities. (b) Intra-abdominal small-cell desmoplastic tumors may present with hepatomegaly. When imaging fails to reveal a primary source, a liver biopsy specimen can show the diffuse infiltration of the parenchyma by ill-defined small cells. The metastases may or may not be accompanied by a desmoplastic reaction. (c) Desmin positivity is a characteristic of these tumors whose prognosis is dismal. (d) Infants may present with megakaryoblastic leukemia with or without stigmata of Down’s syndrome. This 4-month-old girl had marked hepatomegaly and only minimal evidence of hepatocellular dysfunction. The peripheral blood and bone marrow were unremarkable except for subtle fibrosis of the marrow. Initially, the cellularity was not reduced. (e) Immunostaining provided the diagnosis when the platelet glycoprotein B was positive in the tumor cells (Table 14.7)
14.3.1 Imaging and Staging
Careful delineation of the extent of hepatic involvement and metastases is absolutely critical in the management of liver tumors. Staging in the United States combines imaging with surgical judgment about resectability (Fig. 14.16). International groups are united in testing the SIOPEL PRETEXT system at diagnosis (Meyers et al. 2009). Regardless of the system, all groups agree that resection should be performed as early as possible to reduce the need for and extent of chemotherapy (von Schweinitz et al. 1995b; Finegold et al. 2008; Meyers et al. 2011). Imaging performed by CT with contrast and ultrasonography is added to further delineate vascular involvement (Fig. 14.17). The availability of sensitive ultrasound, with Doppler flow studies, readily detects cystic lesions and facilitates fine-needle aspiration for cytology (Fig. 14.18). Magnetic resonance angiography is used for delineating hemangiomas, hemangioendotheliomas, and arteriovenous malformations, as well as preparing the surgeon for dealing with the vagaries of hepatic arterial supply in all attempted resections (McCarville and Kao 2006). Contrast-enhanced 3-D magnetic resonance angiography seems to be the most effective technique for the demonstration of intrahepatic tumor distribution and involvement of portal vein and inferior vena cava, sparing the patient ionizing radiation and possible allergic reactions to dye needed for angiography. Gadolinium serves to shorten the T1 relaxation time of blood, making it brighter than tissues regardless of blood flow. Intraoperative ultrasonography may be useful during hepatectomy in judging the involvement of major vessels (Finegold et al. 2008). Whether these methods will achieve the sensitivity necessary to distinguish hemangiomas/hemangioendotheliomas from HB in infants remains uncertain. As illustrated, the former are often solid and poorly vascularized (Fig. 14.19a) resembling epithelial HB (Fig. 14.19b), whereas many HBs can have large telangiectatic vessels with ample perfusion (Fig. 14.19c). CT is generally used to detect intra-abdominal lymph node and pulmonary metastases.
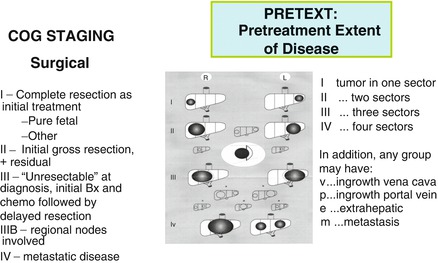
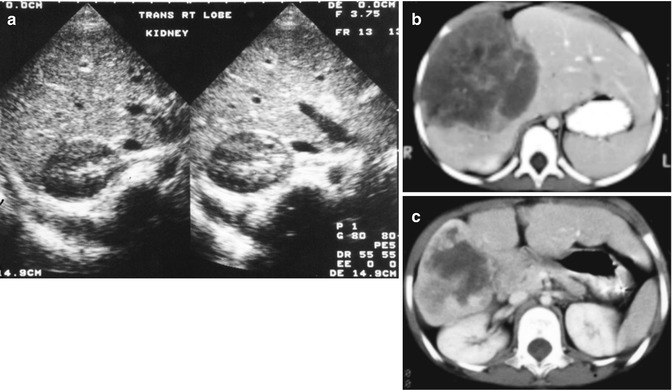
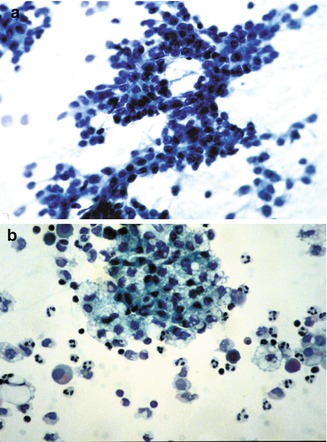
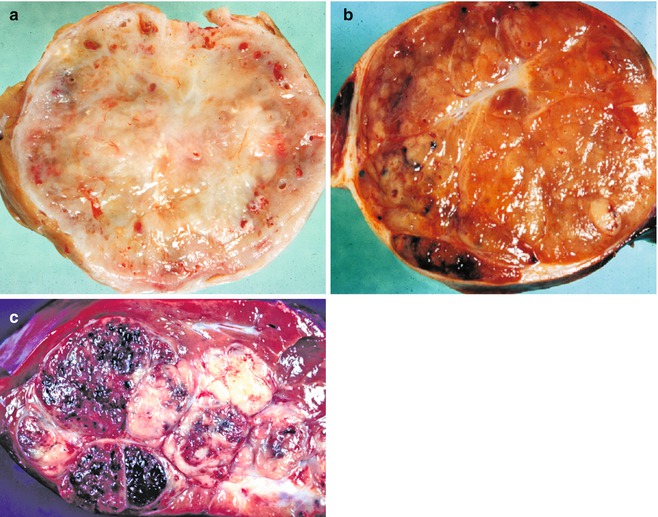
Fig. 14.16
Staging in the United States and the SIOPEL PRETEXT system
Fig. 14.17
Imaging of liver masses. (a) Ultrasound examination of a 4-month-old boy with a symptomatic mass reveals multiple cysts, the largest of which is seen in this image. The smooth linings suggest a benign process, a mesenchymal hamartoma. (b) A computerized tomographic scan of a 3-year-old female with a large mass in the liver shows marked irregularities secondary to abnormal blood flow; a biopsy revealed angiosarcoma (see Fig. 14.37). (c) Following a 3-month course of a multiple-agent chemotherapy described in the text, the patient’s tumor shrank to the point of resectability
Fig. 14.18
Fine-needle aspirate cytology. (a) Embryonal hepatoblastoma showing a high nuclear/cytoplasmic ratio in cells that are moderately cohesive and form cords. (b) Fine-needle aspirate of fetal hepatoblastoma in a 3-year-old child showing abundant lipid droplets in cells that are relatively cohesive and have a relative abundance of cytoplasm. There is an accompanying acute inflammatory infiltrate
Fig. 14.19
Tumors with misleading imaging. (a) An infant girl had a mass in the liver without other symptomatology. Resection revealed a single well-circumscribed mass with modest vascularity. Central depression represents some scarring, but this lesion was a type I hemangio(endothelio)ma with only a small foci of vascular dilatation toward the periphery. Imaging studies led to a clinical diagnosis of hepatoblastoma. (b) The gross appearance of this epithelial form of hepatoblastoma is very similar to the hemangio(endothelio)ma; the imaging studies were entirely comparable. A few dilated vascular spaces are present at the periphery, but the tumor is primarily solid. The tan-orange color reflects excellent differentiation in this fetal hepatoblastoma. (c) This typical mixed hepatoblastoma has extensive hemorrhage secondary to many telangiectatic blood vessels. The gray-white tissue represents abundant stroma in which osteoid-like material was present as well as some poorly differentiated spindle cells. The slightly yellowish area represents foci of necrosis
Dissemination of hepatic malignancies occurs within portal veins (Fig. 14.20a) and follows the expected ready access of infiltration into hepatic veins with frequent lung involvement (Fig. 14.20b). Further spread to the brain may occur (Fig. 14.20c). Hilar lymph node metastases are relatively infrequent, but capsular rupture of subcapsular masses either pre- or intraoperatively can upstage an otherwise resectable malignancy.
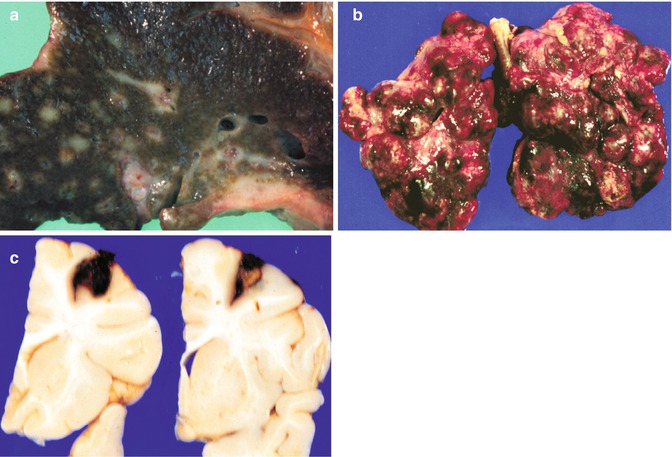
Fig. 14.20
Dissemination of liver tumors. (a) A cross section of a liver at autopsy in a patient with hepatocellular carcinoma shows extensive dissemination via portal veins. (b) The lungs reveal multiple masses of secondary hepatocarcinoma. Similar spread occurs with HB, rhabdoid tumors, embryonal sarcomas, and angiosarcomas. (c) Coronal sections of the brain from a patient with a hepatic rhabdoid tumor reveal metastatic disease
14.3.2 Biochemical Tests and Tumor Markers
Serum AFP is the most useful indicator of hepatocellular neoplasia (Table 14.11). This product of fetal hepatocytes (Fig. 14.21a) is markedly elevated in 80–90 % of HB and 60–70 % of HCC (Lopez-Terrada et al. 2014). Lesser degrees of elevation in infants can be due to variations in the rate of decline after birth or secretion from regenerating hepatocytes adjacent to hemangioendotheliomas (Han et al. 1998) or mesenchymal hamartomas (Boman et al. 2004; Cajaiba et al. 2007). AFP also can be elevated in pancreatoblastomas and yolk sac tumors, which may occur as primary tumors in the liver or together with HB. On the contrary, AFP will not be increased when HBs are primarily composed of the small-cell undifferentiated type, or in most fibrolamellar carcinomas, but even some typical fetal HB have failed to produce detectable increases in serum AFP (Fig. 14.21b). Children with AFP less than 100 ng/ml had poor response to chemotherapy in the German HB study, reflecting the lack of differentiation. This has been amply confirmed by SIOPEL (De Ioris et al. 2008) and in a review of all documented cases with small undifferentiated HB, who failed to respond to usually effective chemotherapy (Trobaugh-Lotrario et al. 2009). Following the AFP level in patients with unresectable HB after chemotherapy has great prognostic value. In a retrospective analysis of 31 patients in the CCG series from 1986 to 1989, a prompt decline of greater than or equal to 2 logs was associated with disease-free survival in 15 of 20 patients, whereas 5 of 6 with no response and 4 of 5 with a delayed or lesser response died from their tumor (Van Tornout et al. 1997). The rapid decline of AFP combined with the degree of shrinkage of tumor by imaging led the Vanderbilt group to propose resection after just two instead of four cycles of chemotherapy, with a corresponding reduction in toxicity (Lovvorn et al. 2010). By using radiolabeled (Tc99m) Fab fragments of monoclonal antibody to AFP as a tracer, Kairemo et al. (2002) were able to successfully transplant the liver of a child who previously had lung metastases. Ascertaining the degree of fucosylation of AFP may provide for early detection of HCC in cirrhotics (Aoyagi et al. 2011). Screening of the population of Shanghai with serum AFP detected subclinical HCC in 263 persons over 33 years (Tang et al. 1993). Unlike patients with symptoms, 81.4 % had resectable tumors (vs. 46.8 %), and 60.5 % of those resected survived 5 years (vs. 36.8 %). However, AFP is elevated in patients with hereditary tyrosinemia type I and ataxia–telangiectasia prior to the development of HCC (Finegold 2004).




Table 14.11
Serum alpha fetoprotein
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
