Histologic features
Neonatal hepatitis
Obstruction
Giant-cell transformation
Usually diffuse
Usually focal
Lobular disarray
Usually marked
Usually mild
Bile duct proliferation
Usually absent, focal ductular reaction can be present
Prominent and widespread
Bile plugs in interlobular ducts
Absent
Present
Portal fibrosis
Late and variable
Early and progressive
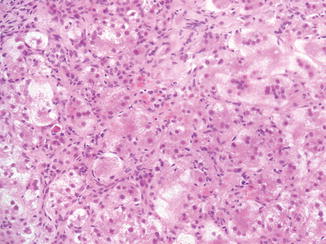
Fig. 12.1
Neonatal hepatitis. Prominent lobular changes characterized by giant-cell transformation of hepatocytes, inflammation, and single-cell necrosis are more widespread than in obstructive processes
12.2.2 Congenital Infections
Congenital infections are acquired transplacentally or at the time of birth (Table 12.2). Some pathogens, such as Cytomegalovirus and Listeria, can affect the fetus following either route, whereas others (rubella, toxoplasmosis) will typically infect the fetus before birth or at the time of delivery (herpes simplex). In general, the earlier the infection, the less active the changes will be at the time of the biopsy. Therefore, most cases of infectious neonatal hepatitis are probably secondary to relatively late infection during the perinatal period.
Table 12.2
Infections that may cause neonatal liver disease
Cytomegalovirus |
Herpes simplex |
Varicella |
Rubella |
Echovirus |
Coxsackie |
Hepatitis B |
Hepatitis C |
Parvovirus |
Listeria |
Mycobacteria |
Syphilis |
Toxoplasmosis |
Although bacteria are a frequent cause of neonatal infections, organ involvement is usually not limited to the liver; percutaneous liver biopsies are rarely performed in infants with sepsis. Viruses are by far the most common cause of infectious neonatal hepatitis.
12.2.2.1 Cytomegalovirus
Cytomegalovirus (CMV) infection is the most common intrauterine viral infection and may result in severe injury to the fetus (Brown and Abernathy 1998; Dollard et al. 2007; Swanson and Schleiss 2013). Forty percent to 50 % of infants delivered to mothers with primary CMV will have congenital infections. Of these, 5–18 % will be overtly symptomatic at birth. The mortality rate in these children is almost 30 %. The most severely affected infants present with growth retardation, microcephaly, hydrocephalus, chorioretinitis, thrombocytopenia, and hepatitis (Becroft 1981). Jaundice and hepatomegaly may develop after the first month of life and may persist for weeks or months. Liver disease ranges from mild portal inflammation with viral inclusions in the bile duct epithelium to severe cholestatic hepatitis. Although portal and intralobular fibroses have been described, there is no evidence that CMV hepatitis progresses to cirrhosis. Kershisnik et al. (1992) reported a case of a fetus with evidence of CMV infection and liver disease with the phenotype of neonatal hemochromatosis. Except for these unusually severe cases, the changes in the liver are characterized by variable cholestasis, prominent hematopoietic activity, focal giant-cell transformation, and characteristic inclusions (Fig. 12.2). CMV infection has also been associated with paucity of the bile ducts (Finegold and Carpenter 1982; Dimmick 1993; Kage et al. 1993; Chiu et al. 1996), and since the viral inclusions are usually seen in the bile duct epithelium, it has been speculated that CMV might be responsible for bile duct damage and even progression to biliary atresia. The potential role of CMV in the development of biliary atresia is further discussed in Chap. 10 of this book. Liver calcification in cases of prenatal CMV infection has been detected radiologically, and pathologically, the deposits are seen in portal spaces and under the capsule (Becroft 1981). Intrapartum infection is less common and severe than transplacental infection. Most postnatal infections are acquired via infected breast milk or blood transfusion.
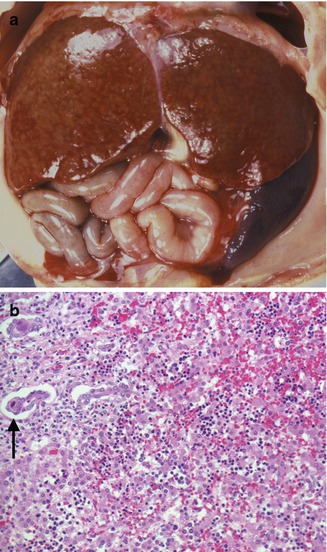
Fig. 12.2
Congenital CMV. A 27-week gestation fetus with hydrops fetalis. Liver with faint nodularity, marked splenomegaly (a), and characteristic inclusions in the ductal epithelium (arrow) (b). Focal hemorrhage and collapse are seen in the parenchyma
12.2.2.2 Herpes Simplex Virus
Herpes simplex virus (HSV) infection is usually acquired when the infant comes into contact with viable virus during intrapartum transit through an infected birth canal (Kimberlin 2005). However, some cases are transmitted after birth, when the infant is exposed to caregivers with orolabial herpes, herpetic whitlow, or lesions in other sites (Stone et al. 1989). Less commonly, infants also may become infected while still in utero, due to either ascending infection from the cervix or vulva or from transplacental transmission (Marquez et al. 2011). The risk to the infant varies with the type of maternal infection and route of delivery. The risk (57 %) is highest for vaginal deliveries in the presence of a symptomatic maternal first episode lesion (Brown et al. 2003). HSV type 2 is responsible for almost 80 % of perinatal infections. Neonatal herpes presents in one of three ways: disseminated, limited to the central nervous system, or limited to the skin and mucous membranes. The least common but most lethal form is disseminated infection, which occurs in approximately 25 % of infected newborns (Kimberlin 2005). The initial symptoms are nonspecific; vesicular lesions are usually absent. Rapid deterioration follows and shock develops towards the end of the first week of life. There is multiorgan system involvement, which predominantly affects the liver and adrenal glands. The hepatic lesions consist of small areas of necrosis diffusely distributed throughout the parenchyma. Grossly these areas are yellow-white, against an intensely red background, and measure from 0.5 to 3.0 mm (Singer 1998) (Fig. 12.3a). Microscopically, the areas of necrosis are usually devoid of inflammatory cells. Although all cellular elements are involved, there is no collapse of the reticulin network. Viral cytopathic effect is seen in the adjacent viable tissues (Fig. 12.3b).
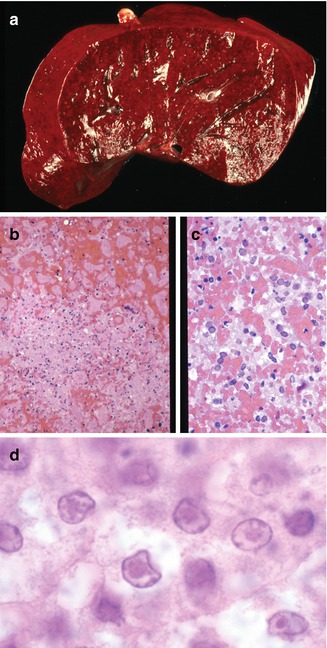
Fig. 12.3
Congenital herpes. Liver with diffuse hemorrhage and punctate areas of necrosis (a). Areas of necrosis have little inflammatory reaction (b). Characteristic viral cytopathic effects including Cowdry type B inclusions (c) and type A inclusions (d) are seen in hepatocytes
12.2.2.3 Echoviral Infections
Echoviral infections leading to acute liver failure occur most frequently in the neonate, typically in the summer and fall (Moore 1982). The clinical course and outcome may vary depending on the type of echovirus; echovirus type 11 is the most commonly mentioned serotype in fatal neonatal infections (Wang et al. 2001). Coagulopathy with disseminated intravascular coagulation and liver failure dominate the clinical picture. Diffuse centrilobular hemorrhagic necrosis with minimal inflammation is the main pathologic finding (Fig. 12.4). Wang et al. (Wang et al. 2001) observed two histologic patterns of liver involvement: intravascular coagulation in the early clinical course and a veno-occlusive component in later stages of the disease. They postulated viral damage to vascular endothelium rather than direct hepatocyte injury as the mechanism of liver injury. Other organs involved in disseminated infection include the brain, heart, and adrenal glands. No viral cytopathic effect is seen by light microscopy.
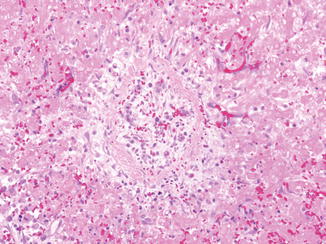
Fig. 12.4
Neonatal echovirus fulminant hepatitis. Centrilobular necrosis with minimal inflammation. The central vein shows obliteration of the lumen by loose fibrous tissue
12.2.2.4 Coxsackie B Virus
12.2.2.5 B19 Parvovirus
B19 parvovirus is a major cause of nonimmune hydrops. Intrauterine infection results in fetal anemia, myocarditis, ocular lesions, and, less commonly, severe liver disease (Metzman et al. 1989; White et al. 1995; Silver et al. 1996; de Jong et al. 2006). The liver disease is characterized by severe hepatocellular siderosis with sinusoidal fibrosis, foci of GCT, bile duct proliferation, cholestasis, and excessive hematopoiesis. Nuclear inclusions are usually not seen in hepatocytes, but they can be identified in erythroid precursors in the liver (Fig. 12.5). In one case (White et al. 1995), extrahepatic siderosis with a hemochromatotic distribution, as seen in neonatal hemochromatosis, was documented. In children and young adults, acute hepatitis and ALF have been associated to B19 parvovirus infection (Yoto et al. 1996; Sokal et al. 1998; Tung et al. 2000; Dame et al. 2002).
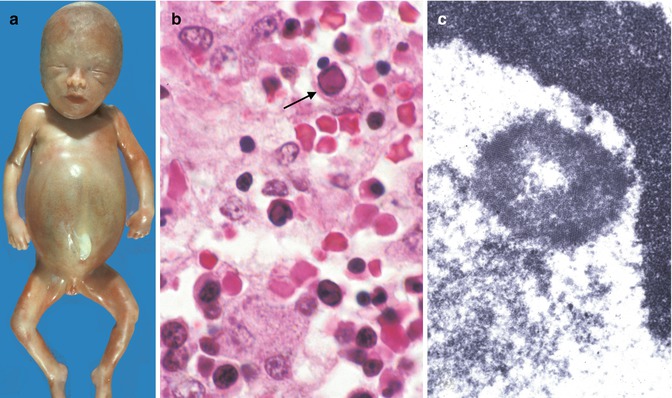
Fig. 12.5
Congenital parvovirus. Hydrops fetalis (a). Section of liver demonstrates viral cytopathic effect in erythroid precursors (arrow) (b). Ultrastructural view of intranuclear viral crystalline material (c)
12.2.2.6 Rubella
Congenital rubella is currently a relatively rare condition due to the widespread use of rubella vaccine. In addition to numerous developmental problems leading to permanent sequelae, congenital rubella frequently causes liver disease (Esterly et al. 1967; Strauss and Bernstein 1968). Early lesions include swelling of hepatocytes, cholestasis, and giant-cell transformation (Fig. 12.6). In some cases, proliferation of bile ducts may suggest extrahepatic biliary obstruction, and biliary atresia leading to biliary cirrhosis has been reported (Rosenberg et al. 1981).
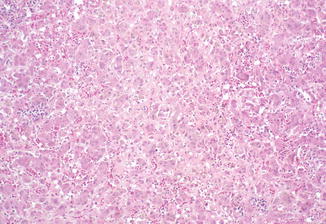
Fig. 12.6
Congenital rubella. Nonspecific giant-cell transformation of hepatocytes, cholestasis, and increased hematopoietic activity
12.2.2.7 Hepatitis A, B, and C Viruses
Hepatitis A does not pose a serious risk to the fetus, unless the mother becomes severely ill (Duff 1998; Borgia et al. 2012). In contrast, perinatal transmission of hepatitis B virus (HBV) occurs in 10–20 % of women who are seropositive for HBV surface antigen. This frequency increases to almost 90 % in women who are positive for both surface antigen and e antigen, in the absence of immunoprophylaxis (Duff 1998; Borgia et al. 2012). Most infants with perinatally acquired hepatitis B are asymptomatic. They typically have high viral load with normal liver enzyme as characterized by the immune-tolerant phase. As children age, chronic hepatitis occurs as they enter into immune-active phase, characterized by elevated liver enzymes and declining viral load. Some may eventually achieve spontaneous seroconversion (loss of HBeAg and development of HBeAb) and become inactive carriers. Acute icteric hepatitis is rare in perinatal infection (Tang et al. 1998) and may progress rapidly to fulminant hepatitis, particularly when the mother is anti-HB e antigen positive (Chang et al. 1987). The frequency of perinatal transmission of hepatitis C virus (HCV) is highly variable, ranging from less than 10 % to as high as 36 % (Granovsky et al. 1998). Risk factors for HCV vertical transmission include high viral load, HIV coinfection, prolonged rupture of membranes, and fetal scalp monitoring (Mast et al. 2005). Infection commonly results in chronic hepatitis, which is usually asymptomatic in early life.
Nonviral infections that may cause severe neonatal liver disease include specific bacteria such as Listeria monocytogenes and Treponema pallidum and parasites such as Toxoplasma gondii.
12.2.2.8 Listeria monocytogenes
Listeria monocytogenes is a gram-positive bacteria which can cause infection of pregnant women (Mylonakis et al. 2002). Intrauterine infection can lead to severe complications such as amnionitis, preterm labor, spontaneous abortion, stillbirth, or infection of the neonate. In the latter, early-onset listeriosis is usually evident in the infant at the time of birth with meconium staining, jaundice, sepsis, and/or pneumonia. Late-onset listeriosis may occur anytime from 1 to 8 weeks of life. Meningitis is seen in 95 % of the cases (Bortolussi 1990). The liver shows miliary microabscesses that contain abundant bacteria (Fig. 12.7). The lesions develop a granulomatous appearance as mononuclear cells replace polymorphonuclear cells in the abscesses.
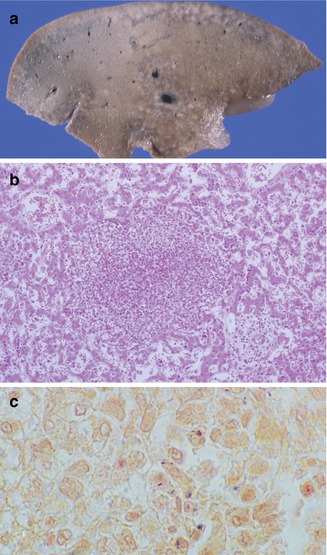
Fig. 12.7
Listeriosis. Cut surface of liver demonstrates miliary microabscesses (a). Microabscess (b) with intracellular and extracellular gram-positive bacilli (c)
12.2.2.9 Congenital Syphilis
Congenital syphilis results from transplacental transmission of Treponema pallidum. The severity of liver disease ranges from mild nonspecific hepatitis to fulminant hepatic failure. Nonspecific findings include portal inflammation, excessive hematopoiesis, and giant-cell transformation, which may be accompanied by areas of necrosis with a miliary distribution containing numerous spirochetes (Fig. 12.8). More commonly, the inflammation is diffuse with compression of the sinusoids and liver cells. Ultrastructurally, spirochetes are localized in the space of Disse (Brooks and Audretsch 1978), which explains the sinusoidal pattern of the inflammation. Only occasional organisms are seen within hepatocytes. These changes are followed by a characteristic diffuse sinusoidal fibrosis with attenuation of the trabeculae (Oppenheimer and Dahms 1981).
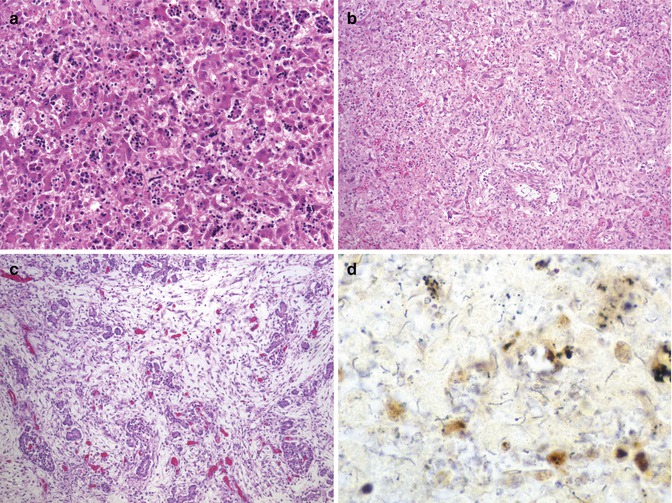
Fig. 12.8
Congenital syphilis. A 4-day-old term neonate with increased hematopoietic activity and inflammation in widened sinusoids (a). Inflammatory activity progresses to sinusoidal fibrosis with attenuation of trabeculae (b). Pancreas with massive interstitial fibrosis and parenchymal obliteration (c). Warthin-Starry stain demonstrates numerous spirochetes in focus of necrosis in the liver (d)
12.2.2.10 Toxoplasmosis
Toxoplasmosis occurs by transplacental transfer of the organism from mother to fetus following acute maternal infection (Lynfield and Guerina 1997; Petersen 2007). The severity of the fetal disease varies inversely with the gestational age at which maternal infection occurs. Without treatment, most fetuses infected early in pregnancy either die or have severe neurologic and ophthalmologic disease. Conversely, nearly all fetuses infected in the second and third trimesters have mild or subclinical disease. Liver damage may be clinically marked in the generalized form, although pathological findings may be mild (Dische and Gooch 1981). Microscopic findings are usually nonspecific, from minimal changes to focal or diffuse hepatitis (Fig. 12.9). Excessive hematopoiesis is common. Cholestasis may be marked. Periportal fibrosis and cirrhosis in a newborn have also been recorded. Giant-cell transformation of hepatocytes is variable. Toxoplasma organisms are rarely present, although they have been noted in Kupffer cells, endothelium, and necrotic foci. Calcifications of necrotic lesions can be seen by image studies.
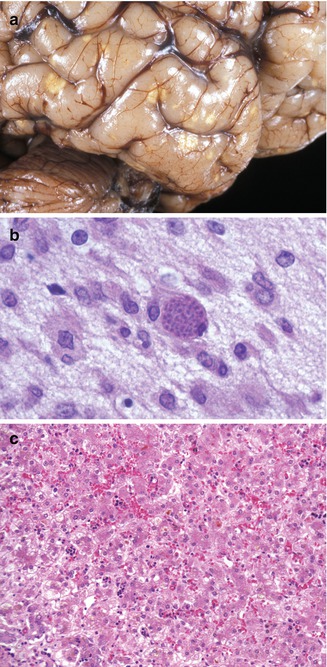
Fig. 12.9
Congenital toxoplasmosis. A 3-week-old neonate with characteristic focally calcified yellow-white lesions in the cerebral cortex (a). Toxoplasma cyst in the brain (b). Changes in the liver are usually nonspecific and include persistent hematopoiesis and cholestasis (c)
12.3 Acute Liver Failure
Acute liver failure (ALF), also called fulminant liver failure, is a rare but life-threatening event in infants, children, and adults. In adults, ALF is defined as the onset of altered mental status (hepatic encephalopathy) and coagulopathy (prolonged prothrombin time) occurring within 8 weeks of initial symptoms of a hepatitis-like illness in a person without prior liver disease (Ostapowicz and Lee 2000; Gill and Sterling 2001; Ostapowicz et al. 2002). Defining ALF in children is more difficult due to problems in assessment of encephalopathy in infants and children and to the frequency of occult chronic liver disease in this age group (Squires 2008). Recently the Pediatric ALF study group, a multicenter consortium collecting clinical data and serum samples prospectively from children with ALF, has adopted criteria for the diagnosis of ALF in children (Table 12.3).
Table 12.3
ALF diagnostic criteria (pediatric ALF study group)
1. Evidence of a liver-related illness |
2. No history of known chronic liver disease |
3. Coagulopathy (uncorrected by vitamin K): |
(a) INR ≥ 1.5 PLUS encephalopathy OR |
(b) PT ≥ 15 PLUS encephalopathy OR |
(c) INR ≥ 2.0 WITH or WITHOUT encephalopathy OR |
(d) PT ≥ 20 WITH or WITHOUT encephalopathy |
12.3.1 Differential Diagnosis
ALF can have many causes ranging from infections (particularly viral) to toxic exposures to specific metabolic diseases, though many cases are still idiopathic despite extensive evaluation (Durand et al. 2001; Ostapowicz et al. 2002; Squires 2008; Devictor et al. 2011; Shanmugam et al. 2011). Though viral causes are the most common identified overall, other causes vary by age (Devictor et al. 2011; Shanmugam et al. 2011) (Table 12.4). In children, the known causes of ALF are more varied, yet the proportion of idiopathic cases is much higher, greater than 40 % (Black 2009). There is a considerable contribution, perhaps underestimated in most series, from a variety of fatty acid oxidation defects and other metabolic diseases, particularly in young infants (Durand et al. 2001; Lee and Sokol 2007; Sze et al. 2009).
Table 12.4
Common causes of ALF
Infants | Children and adolescents |
|---|---|
Viral infections | Viral infections |
Herpes | HAV, HBV, HCV, HEV |
Echovirus | EBV |
Coxsackie virus | Varicella |
CMV | Adenovirus |
Immune | Drug/toxin |
Gestational alloimmune liver disease | Dilantin |
Metabolic | Valproate |
Tyrosinemia | Acetaminophen |
Galactosemia | Isoniazid |
Hereditary fructose intolerance | Amanita |
Mitochondrial diseases | Immune |
Vascular | Autoimmune hepatitis |
Asphyxia | Hemophagocytic Histiocytosis |
Congenital heart disease | Metabolic |
Idiopathic | Wilson’s disease |
Mitochondrial diseases | |
Malignant hyperthermia | |
Vascular | |
Budd-Chiari syndrome | |
Veno-occlusive disease | |
Idiopathic |
A total of 28 liver transplants were performed for the diagnosis of ALF at the Children’s Hospital of Philadelphia between January 1, 1998 and December 31, 2012. Of those cases, in 12 no diagnosis was identified; 5 cases were caused by documented viral infection (enterovirus 2, echovirus 1, and influenza virus 2); 5, Wilson’s disease; 1, tyrosinemia; 3, mitochondrial disease; and 2, autoimmune hepatitis. A specific cause for ALF may be more frequently identified in young infants; in this same series, a diagnosis was made in 4 of 6 patients under 3 months old at presentation (2 tyrosinemia, 1 echovirus, 1 familial erythrophagocytosis). It is important to note that in this small series, only transplantation has been reviewed. For this reason, there is considerable ascertainment bias as the unknown proportion of children who died without transplant or recovered may have a distinct set of underlying diagnoses.
12.3.2 Clinical Features
Infants and children with ALF have a diverse presentation depending on age, underlying diagnosis, and timing of presentation (Devictor et al. 2011). Young infants may present with sepsis-like syndromes within the first few days of life, cholestatic jaundice, or bleeding diathesis. ALF in newborns and very young infants is of particular interest in that several disorders can cause ALF in this age group and are not seen in older infants or children (Bhaduri and Mieli-Vergani 1996; Durand et al. 2001; Whitington et al. 2001).
The presentation of ALF in older children often begins with a prodromal illness followed by development of jaundice and other features of acute hepatitis, including abdominal pain and vomiting. In later stages, children may experience epistaxis or other sources of bleeding due to the development of coagulopathy, following which there may be gradual or rapid onset of hepatic encephalopathy. Frequent physical examination and mental status checks are important in monitoring the degree of hepatic dysfunction, the potential for recovery, and the need for intensive care. A rapidly shrinking liver with falling enzymes can be the harbinger of complete parenchymal collapse and urgent need for transplantation (Bhaduri and Mieli-Vergani 1996; Whitington et al. 2001).
Laboratory findings of elevated liver enzymes, bilirubin, and prothrombin time are characteristic. Liver enzymes may be dramatically elevated at presentation (over 10,000 in many cases), and falling enzymes may represent recovery or worsening disease as the entire liver parenchyma is lost. These situations may be differentiated by the prothrombin time, which will improve in recovery states but worsen in end-stage failure. Thrombocytopenia may be the result of occult underlying chronic liver disease (due to splenomegaly and sequestration) or may be due to acute viral infection. Specific diseases are associated with some characteristic laboratory findings; however, these are not always present and are not pathognomonic. For example, Wilson’s disease may be associated with acute hemolysis due to the oxidant effect of copper released from the collapsing liver. Other findings in Wilson’s disease include Fanconi-type aminoaciduria; however, this can be seen in a variety of clinical settings causing acute liver disease. Disproportionate acidosis may be an indicator of metabolic liver disease as is the history of mild developmental delay or hypotonia.
While metabolic diseases that may cause ALF are discussed in Chap. 13 of this book, infections and toxin exposures as well as other specific etiologies of ALF are discussed here.
12.3.3 Pathology
Most cases of ALF include a significant degree of hepatocellular necrosis. The necrosis can be massive (entire liver), submassive (major portions of the liver), zonal (same region of hepatic lobules), or random (nonzonal). The degree of inflammation is variable, and when present, a particular type of inflammatory cell may predominate. Although in most instances the histopathologic features are not specific enough to identify a particular etiology, some findings may suggest a narrower gamut of possible causes (Table 12.5). Drug-induced liver damage is often indistinguishable from acute viral hepatitis. Eosinophils or granulomas may suggest a drug reaction, but they are no proof of drug etiology nor necessary for the diagnosis (Scheuer and Lefkowitch 2000). Idiosyncratic drug injury (unpredictable, not dose-dependent) usually causes nonzonal necrosis, but exceptions such as halothane toxicity, which causes zonal necrosis, do exist. Conversely, intrinsic toxins (predictable, dose-dependent), such as acetaminophen, usually cause zonal necrosis. Autoimmune hepatitis may present clinically as ALF with histological evidence of marked bridging necrosis and extensive collapse (Fig. 12.15). Portal and lobular inflammation are usually prominent and may include a large number of plasma cells (Czaja and Carpenter 1993). Giant-cell transformation of hepatocytes in an older child is highly unusual and may suggest an autoimmune etiology. ALF can be the first manifestation of Wilson’s disease. Histologically, massive necrosis is characteristic, and evidence of nodular regeneration and fibrous septa is usually present (McCullough et al. 1983; Davies et al. 1989), indicating that histologic lesions develop before the disease is clinically apparent. Features suggestive of Wilson’s disease such as Mallory bodies, glycogenated nuclei, and focal steatosis should be sought in the remaining viable parenchyma.
Table 12.5
Histologic findings and differential diagnosis of ALF
Histologic findings | Possible causes | ||
|---|---|---|---|
Necrosis | Inflammation | Other | |
Random | Minimal | Viral cytopathic effect | Herpes simplex |
Varicella zoster | |||
Adenovirus | |||
Centrilobular | Minimal | Echovirus | |
Coxsackie virus | |||
Variable steatosis | Acetaminophen | ||
Halothane | |||
Valproate | |||
Canalicular cholestasis | Drugs | ||
Numerous eosinophils | |||
Granulomas | |||
Bridging | Numerous plasma cells | Autoimmune | |
Massive | Variable | Hepatitis A–C | |
Non-A–G hepatitis | |||
Isoniazid | |||
Phenytoin | |||
Mallory bodies | Wilson’s disease | ||
Patchy steatosis | |||
Glycogenated nuclei | |||
12.3.4 Gestational Alloimmune Liver Disease (GALD)
Gestational alloimmune liver disease has been established as the cause of fetal liver injury resulting in nearly all cases of neonatal hemochromatosis (Whitington 2012). Neonatal hemochromatosis is a clinical syndrome in which severe fetal liver injury causes iron overload due to poor regulation of maternofetal iron flux. GALD has been shown to be the result of maternofetal alloimmune liver injury characterized by subacute or chronic liver disease with associated iron overload at extrahepatic sites in utero. This entity is discussed here because it typically presents clinically as ALF at birth or shortly afterwards. Profound coagulopathy, thrombocytopenia, hypoalbuminemia, hypoglycemia, and low transaminases are common laboratory findings (Goldfischer et al. 1981; Knisely et al. 1987; Knisely 1992). It has also been reported in fetal hydrops and stillbirths (Wisser et al. 1993). The outcome is usually poor, but survival is reported (Muller-Berghaus et al. 1997; Sigurdsson et al. 1998). Ferritin and iron levels are quite elevated, but these laboratory findings are not diagnostic (Knisely 1992). Documented iron deposition in peripheral tissues is diagnostic in this clinical setting and can be shown histologically in the buccal salivary glands by a minimally invasive biopsy (Knisely et al. 1988) or by magnetic resonance imaging of the pancreas (Oddone et al. 1999). Iron overload may potentiate damage initiated by other agents. Its accumulation has been noted to be reversible rather than progressive, yet the trials of chelation therapy have not improved outcomes (Jonas et al. 1987; Knisely 1994; Muller-Berghaus et al. 1997; Sigurdsson et al. 1998). More recently this disorder has been shown to be the result of maternal antibody against an as-yet-unidentified fetal liver antigen (Whitington 2012). Infants have been successfully treated with exchange transfusion (analogous to management of fetal hydrops due to Rh incompatibility), and recurrence of disease has been prevented by maternal gestational therapy with intravenous IgG (analogous to treatment with Rhogam for Rh disease) (Rand et al. 2009). Thus, the condition does not appear to be hereditary but has a pattern of recurrence which is similar to other maternofetal alloimmune diseases (Whitington 2012). Recurrence risk without treatment can be over 90 % (Whitington and Kelly 2008). The liver pathology is characterized by extensive hepatocellular loss accompanied by diffuse fibrosis and varying degrees of cholestasis (Knisely et al. 1987; Silver et al. 1987) (Fig. 12.10). Surviving hepatocytes may show giant-cell or pseudoglandular transformation. Central vein sclerosis with occlusion of the lumen by fibrous tissue has been described (Knisely 1992). Hemosiderin deposition, in a hemochromatotic distribution, is usually marked in hepatocytes, ductular structures, and bile duct epithelium. More characteristic, however, is the extrahepatic iron deposition with reticuloendothelial sparing in the parenchyma of multiple organs including the thyroid, pancreas, adrenals, heart, kidneys, and glands of upper respiratory and digestive tracts. Since hepatic parenchymal iron overload can also be observed in Zellweger syndrome, galactosemia, tyrosinemia, and many viral infections (Murray and Kowdley 2001), these conditions should be distinguished on clinical and laboratory grounds. Hepatic iron concentration in patients with GALD is increased and measures between 240 and 38,200 μg/g dry liver weight (average in the neonate is less than 250 μg/g), but it does not distinguish the diagnosis from other forms of liver disease (Silver et al. 1993). There is no evidence that GALD is related to hereditary hemochromatosis (Kelly et al. 2001). Chelation therapy has been used empirically when the disease was thought to be the result of iron overload injury, but it has been repeatedly shown to be ineffective and is no longer recommended. Liver transplant is an alternative if medical therapy has failed (Jonas et al. 1987; Sigurdsson et al. 1998). Recently, cases of GALD causing acute liver failure and fetal demise without the neonatal hemochromatosis phenotype have been reported (Whitington et al. 2011). Rare non-GALD causes of neonatal hemochromatosis include bile acid synthetic disorders (Shneider et al. 1994) and mitochondrial DNA depletion syndromes (Hanchard et al. 2011).
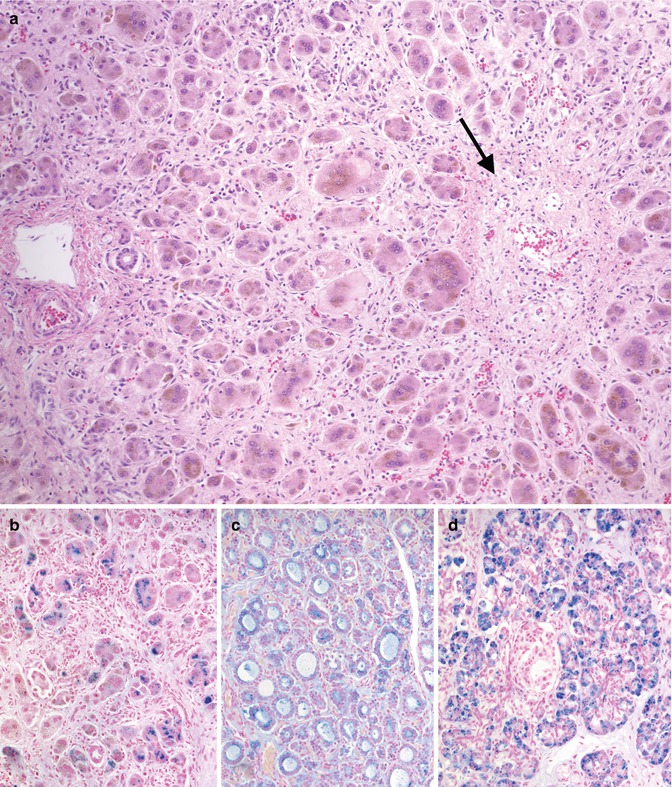
Fig. 12.10
Gestational alloimmune liver disease. Diffuse pericellular fibrosis encircling residual hepatocytes with giant-cell transformation (a). A central vein shows fibrous occlusion of the lumen (arrow). Iron stain demonstrates heavy deposition in hepatocytes (b), thyroid follicular cells (c), and exocrine pancreas (d)
12.4 Chronic Hepatitis
The most common causes of chronic hepatitis in children are viral hepatitis and autoimmune hepatitis (Table 12.6). The histologic features may favor one or another cause of chronic hepatitis and are important to exclude other causes of liver disease and evaluate the severity of the lesion. Less frequently, a special stain or immunohistochemical stain may positively establish a diagnosis. The histologic features of chronic hepatitis can be divided into three anatomical sites: portal, interface, and lobular. Generally, they include two types of lesions: necroinflammation and fibrosis. Evaluation of the severity of these lesions constitutes the basis for the various scoring systems, which are used for both pediatric and adult patients (Scheuer 1991; Batts and Ludwig 1995; Ishak et al. 1995). None of these changes are specific for any particular etiology, and a diagnosis is established by a combination of clinical, laboratory, and pathological findings. Chronic hepatitis may be difficult to differentiate from acute viral hepatitis, particularly when the inflammatory activity is severe. Centrilobular or bridging necrosis, lobular inflammation, and cholestasis are more predominant in acute viral hepatitis. Stains for elastic fibers may help in the distinction between collapse secondary to bridging necrosis and fibrous septa of chronic disease. Elastic fibers accumulate in the latter but are not present in recent collapse (Scheuer and Maggi 1980).




Table 12.6
Histologic differential diagnosis of chronic hepatitis in children
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
