Usual presentationa
Disorder
Additional clinical features
Inheritance
Prenatal/neonatal
Megacystis-microcolon hypoperistalsis syndrome
Markedly enlarged bladder, intestinal malrotation, short dysmotile colon; females more common than males
Probably autosomal recessive
Hirschsprung disease
20 % syndromic (other malformations) and 80 % nonsyndromic (isolated); males more common than females
Multigenic
Congenital hypoganglionosis
None established to date
Intestinal neuronal dysplasiab,c
Frequently associated with other primary causes of dysmotility (e.g., Hirschsprung disease)
None established to date
X-linked intestinal pseudo-obstruction/diffuse abnormal layering of small intestinal smooth muscle
Intestinal malrotation, short small intestine, high rates of spontaneous fetal demise and various malformations
X-linked recessive (males)
Retarded neuronal maturation
None established
Cystic fibrosis
Meconium ileus or peritonitis; chronic lung infection; pancreatic insufficiency
Autosomal recessive
Atrophic desmosis
None established
Childhood/adolescence
Diffuse ganglioneuromatosis
Multiple endocrine neoplasia type 2B
Neurofibromatosis type 1
Cowden syndrome
Autosomal dominant
Neuronal nuclear inclusion disease
Ataxia, dysautonomia
Autosomal dominant
Mitochondrial disorders
Severe constipation, abdominal distension, vomiting; encephalopathy, myopathy, ophthalmoplegia, stroke-like episodes
Autosomal recessive or mitochondrial
Familial visceral myopathy
Abdominal distension, segmental or generalized intestinal dilatation (e.g., megaduodenum)
Autosomal dominant or autosomal recessive
Sporadic visceral myopathy
Similar to familial visceral myopathy
None established
Myotonic dystrophy
Skeletal muscle weakness
Autosomal dominant
Duchenne muscular dystrophy
Skeletal muscle weakness and atrophy
X-linked recessive
Young adults (or older)
Connective tissue disorders; paraneoplastic inflammatory ganglionitis; systemic neurodegenerative disorders (e.g., spinal cerebellar ataxia); Chagasic megacolon
Many patients with enteric motor defects undergo repeated surgeries to exclude physically obstructive lesions, obtain diagnostic biopsies, and/or empirically resect portions of bowel, in an effort to improve quality of life. The surgical pathologist plays an important role in the diagnosis and management of these patients by narrowing the differential diagnosis to a subgroup or single etiology, or at least excluding recognizable disorders (e.g., Hirschsprung disease) that require surgical management. For pathologists, it is helpful to classify anatomically distinct forms of pseudo-obstruction as shown in Table 7.2. This table is based loosely on the London Classification, which was developed by consensus among an international group of pathologists, who subdivided adult and pediatric histopathological phenotypes into those considered “etiological” (proximal causes of intestinal dysmotility) versus “morphological associations” that may or may not contribute directly to dysmotility per se (Knowles et al. 2010). The same body also proposed guidelines for handling and interpretation of gastrointestinal tissue samples from patients with dysmotility (Knowles et al. 2009). The London Classification provides a useful framework for considering histopathological patterns of neuromuscular disease, while recognizing that individual disease processes may be characterized by one or more pattern (e.g., lymphocytic ganglionitis and acquired hypoganglionosis in Chagasic megacolon) or involvement of both neural and muscular tissues (e.g., mitochondrial disorders).
Table 7.2
Pathological classification of pediatric gastrointestinal neuromuscular pathology
Neuropathies |
Aganglionosis (Hirschsprung disease) |
Hypoganglionosis |
Congenital |
Acquired |
Hyperganglionosis |
Ganglioneuromatosis |
Intestinal neuronal dysplasia |
Retarded neuronal maturation |
Gliopathies |
Myopathies |
Muscular malformations |
Diffuse abnormal layering of small intestinal smooth muscle |
Segmental additional smooth muscle coat |
Focal absence of smooth muscle |
Degenerative visceral myopathy |
Familial |
Sporadic |
Inflammatory |
Visceral involvement in muscular dystrophies |
Combined |
Mitochondrial disorders |
Megacystis-microcolon-intestinal hypoperistalsis syndrome |
Colonic desmosis |
Idiopathic (no diagnostic pathology) |
7.2 Enteric Neuropathies
7.2.1 General Concepts
Enteric neurons and glia are derived from neural crest cells that populate the gut during embryogenesis (Newgreen and Young 2002a, b). The major source of ganglion cell precursors arises from crest in the hindbrain region and disseminates rostrocaudally through the gut wall to colonize the entire length of the gastrointestinal tract (Young et al. 1998; Burns et al. 2009). In humans, neural precursors enter the foregut in the 4th week after conception and reach the rectum between weeks seven and eight (Okamoto and Ueda 1967; Fu et al. 2003, 2004). A smaller contribution of neural precursors originates from the sacral crest and colonizes the hindgut during the same developmental period (Burns et al. 2009). As neuroglial precursors populate the gut and throughout most of gestation while the gut grows, a subset of these cells continues to replicate (Newgreen et al. 1996; Landman et al. 2007). In animal models and possibly humans, neural precursors continue to arise for a short period after birth, and it has been speculated that excess neurons are eliminated by apoptosis during postnatal maturation of the enteric nervous system, although an unconventional caspase-independent mechanism may be responsible (Pham et al. 1991; Gianino et al. 2003; Uesaka et al. 2007; Enomoto 2009).
The regional density and distribution of enteric neurons and glial cells are determined by programmed developmental events but are also influenced by environmental factors that may impact on survival, differentiation, or replication of neurons at any pre- or postnatal stage. It is possible to isolate cells from dissociated intestinal wall and derive pluripotent stem cells in vitro with the potential to form enteric neurons and glia in vivo (Hotta et al. 2009). Preliminary evidence suggests that enteric neural stem cells express the transcription factor SOX2 and may be glial cells that reside in enteric ganglia (Heanue and Pachnis 2011; Joseph et al. 2011; Laranjeira et al. 2011). Replication and/or differentiation of resident neural stem cells may underlie putative acquired forms of hyperganglionosis (see below), and manipulation of such cells may serve as the basis for future therapies for aganglionosis/hypoganglionosis.
7.2.1.1 Counting Neurons
Accurate quantification of enteric neurons or other cellular components in the enteric nervous system is challenging and the physiological significance associated with mild-to-moderate changes in cell density is unknown. An International Working Group reviewed the literature in this field and concluded that published data are “insufficient in almost every respect and no robust normal ranges can be produced [due to] a disappointing lack of concordance between observations of different investigators” (Knowles et al. 2011). The consensus was that prudent application of existing norms allows for recognition of severe alterations in the density of ENS components, which “would be qualitatively evident to any experienced pathologist.” In reference to the disparate nature of published normative data, several important points must be considered.
1.
Most types of analyses evaluate neural density, not absolute neural numbers. In general, most pathologists examine histological sections or, rarely, whole-mount preparations that represent a small portion of the gastrointestinal tract. The number of neurons in such a preparation may be meaningless unless the size of the tissue sample is considered. Published data usually are expressed as number of neurons per mm of section length or cm2 of whole-mounts. The density of neurons is not necessarily adequate to assess changes in total neuronal number. Many variables, including organ distension and shrinkage (due to fixation and embedding), influence neuronal density without a net gain or loss of neurons.
2.
Criteria used to recognize neurons in histological sections differ and are influenced by section quality, staining, neuronal maturation, and interobserver variability. Some published counting methods include neurons only if their nuclei are in the plane of section. Others rely on recognition primarily of cytoplasmic features of neurons. Immature neurons are difficult to distinguish from glia because they lack the abundant cytoplasm and prominent nucleoli that typify mature ganglion cells.
3.
Neural counts are influenced by section thickness and the distance between sections. Thicker sections sample more tissue and inflate neural density values. Variability is also due, in part, to the irregular distribution of ganglia and the fact that the separations between ganglia are more than the thickness of most histological sections. More accurate estimates can be obtained by evaluating step sections at appropriate intervals to avoid recounting individual cells, or by whole-mount technique, which is impractical for routine use (Heinicke et al. 1987; Smith 1993; Krammer et al. 1994; Karaosmanoglu et al. 1996).
4.
Neuronal densities vary in different parts of the gastrointestinal tract and at different ages. In general, the density of myenteric neurons is less in the small intestine than in the large intestine. However, even within each organ, regional differences in neural density can be significant. In addition, neural densities in the myenteric plexus appear to decline with age (Knowles et al. 2011), although in infants and children the reduction is relatively small and may relate more to growth of the gut than neuronal loss (Smith 1993).
Given all these considerations, it is not surprising that published normative data regarding neuronal densities is highly variable (Table 7.3). The range of normal values is so great that it is improper to reference control data that was collected by others for comparison with counts from patient samples. Instead, it is best to endure the laborious process of establishing normative data from control tissues encountered in one’s own practice or refer cases to a reference laboratory that specializes in such counts (Knowles et al. 2011).
Table 7.3
Normative values for neuronal density in the human ileum and colon based on counts from H&E-stained paraffin sections
Reference | Age | Section thickness (μm) | Ganglion cells/mm | |
|---|---|---|---|---|
Ileum | Colon | |||
Meier-Ruge et al. (1970) | ns | 20 μm (frozen) | 76 | |
Meier-Ruge et al. (1999) | 1–2 years | 15 μm (frozen) | 14.9 ± 3.6 | |
Scharli and Sossai (1998) | 1–16 years | 15 μm (frozen) | 10.0 ± 3.2 | |
Smith (1993) | 4 weeks–10 years | 3 μm | 4 | 7 |
Iwase et al. (2005) | 20–84 years | 4 μm | 3.2 ± 1.0 | |
Ikeda et al. (1988) | ns | 3 μm | >3 | |
Faussone-Pellegrini et al. (1999) | 45–60 years | 4 μm | 1.5–3.5 | |
Amiot et al. (2009a) | 41–87 years | 3–4 μm | 1.5–3.0 | 1.5–3.0 |
Yu et al. (2002) | 17–66 years | 5 μm | 0.93–4.27a | |
Krishnamurthy et al. (1993) | 3 days–11 months | 4 μm | 0.46–0.64a | 0.43–0.80a |
de Souza et al. (1993) | 20–40 years | ns | 0.6b | |
7.2.1.2 Special Approaches
Specialized approaches have been developed to quantify enteric neurons (Table 7.4). These are used primarily by consult services or research groups because the necessary time, equipment, and/or technical expertise cannot be justified with the small volume of suitable specimens encountered in most practices. The simplest approach involves visual counts of ganglion cells in appropriately spaced 3–5 μm-thick, H&E-stained sections from paraffin blocks. Criteria for identifying ganglion cells are defined arbitrarily, but must be applied consistently. Smith used this method to establish normative data for infants and children and to demonstrate the orientation of sections relative to the gastrointestinal tract (transverse vs. longitudinal) did not affect the quantitative data (Smith 1993). Variations of this approach utilize antibodies or histochemical reactions to visualize neurons in paraffin or frozen sections.
Table 7.4
Methods to count neurons
Method | Availability | Supply costs | Labor | Discrimination | Paraffin blocks? | Handling | Reference |
|---|---|---|---|---|---|---|---|
Histological sections | |||||||
H&E | Excellent | Low | Low | Subjective | Yes | Routine | |
Antibodies | Excellent | Moderate | Moderate | Varies | Yes | Routine | Karaosmanoglu et al. (1996) |
Histochemistry (e.g., LDH) | Excellent | Moderate | Moderate | Good | No | Frozen sections | Meier-Ruge et al. (1995b) |
Whole-mounts | |||||||
Cuprolinic blue | Fair | Low | High | Good | No | Manual dissection | Karaosmanoglu et al. (1996) |
Acridine orange | Fair | Low | High | Fair | No | Manual dissection | Sarnat et al. (1985) |
NADPH diaphorase | Fair | Low | High | Excellent | No | Manual dissection | Nemeth et al. (1986) |
Antibodies | Fair | Moderate | High | Excellent | No | Manual dissection | |
Silver stain | Poor | Moderate | High | Excellent | No | Vibratome sections | Schuffler and Jonak (1982) |
Morphometrics | Poor | Moderate | High | Varies | Yes | With antibody or histochemistry | Kubota et al. (2001) |
Meier-Ruge and Longo-Bauer (1997) | |||||||
Alternative methods are based on whole-mount preparations of myenteric, or rarely submucosal, plexus. Such preparations require a more generous biopsy than microscopic sections, usually a full-thickness specimen that includes a minimum of 1 cm2 of the serosal surface. Before or after fixation, the mucosa, submucosa, and circular layer of the muscularis propria are manually dissected from the luminal surface of the biopsy, leaving behind the myenteric plexus, longitudinal muscle layer, and serosa (Tam and Boyd 1990; Ganns et al. 2006). A dissection microscope is used and some practice is required to become proficient at the technique. After fixation, the myenteric plexus with attached tissues is immersed in histochemical reagents or permeabilized and immunolabeled. Finally, the specimen is mounted flat on a glass slide and examined with a compound microscope. Methods used to specifically stain some or all neurons in whole-mount preparations include histochemical and immunohistochemical reagents (Table 7.4, Fig. 7.1).
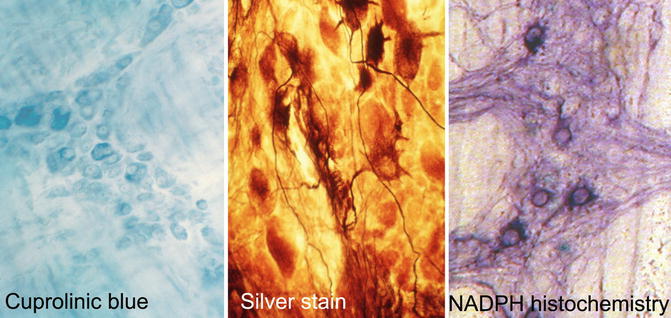
Fig. 7.1
Methods to identify and count neurons in whole-mount preparations of the myenteric plexus. Cuprolinic blue stains the cytoplasm (RNA) of all neurons. Silver stain discriminates argyrophilic (darkly stained) from argyrophobic (pale) neurons and resolves the cytoarchitecture of axons and dendrites. NADPH histochemistry imparts a dark purple color to the cytoplasm of inhibitory motor neurons and interneurons that express neuronal nitric oxide synthase
A somewhat laborious, but morphologically informative, method to image myenteric neurons is Smith’s silver stain technique (Fig. 7.1) (Schuffler and Jonak 1982). The procedure requires several days, including a prolonged period of fixation, and can be modified to specifically enhance resolution of different parts of enteric neurons such as the perikaryon or neurites. Usually, a freezing microtome is used to prepare 50 μm-thick sections parallel to the plane of the myenteric plexus, which contain entire ganglia. Richardson provided a detailed description of this method and modifications that can be made to selectively enhance staining of perikarya versus neurites (Richardson 1960). Although the basis for differential staining of argyrophilic versus argyrophobic neurons with this technique is poorly understood, alterations in the density of neurons of each type have been reported in patients with some clinically distinct forms of pseudo-obstruction (Tanner et al. 1976; Krishnamurthy et al. 1985; Navarro et al. 1990). In addition, the cytoarchitectural details revealed by this method enable subtle alterations in neuronal morphology (e.g., “blunt” dendrites) to be recognized and possibly correlated with impaired peristalsis.
7.2.2 Aganglionosis (Hirschsprung Disease, HSCR)
7.2.2.1 Etiology
Hirschsprung disease (HSCR) refers to a congenital malformation of the enteric nervous system in which the only obligate diagnostic feature is absence of ganglion cells in the myenteric and submucosal plexuses of the terminal rectum. HSCR is traditionally segregated into short- and long-segment types based on whether aganglionosis extends respectively distal or proximal to the splenic flexure. However, distinguishing short-segment disease as that limited to the rectosigmoid colon (~80 % of patients) is more relevant to underlying genetic and epidemiological risk factors (Badner et al. 1990). Rectosigmoid disease is 5.5 times more common in males and less likely to recur in siblings and demonstrates genetic segregation consistent with multifactorial or recessive inheritance. By contrast, aganglionosis that extends proximal to the sigmoid colon shows a significantly lower male gender bias, higher recurrence rate, and genetic properties suggestive of dominant inheritance with incomplete penetrance. Approximately 5 % of patients suffer from total colonic aganglionosis, and rarely the entire small intestine (+/− stomach) is affected.
HSCR is the consequence of incomplete colonization of the gastrointestinal tract by enteric neural crest cells, which originate from the cranial and sacral portions of the neural crest and normally populate the entire length of the bowel by the seventh postconception week of gestation (Fu et al. 2003, 2004; Wallace and Burns 2005). Although environmental factors may influence penetrance or the proximal extent of aganglionosis, mutation of one or more HSCR susceptibility gene is considered to be the basis for the morphogenetic defect.
Hypoganglionosis (paucity, but not complete loss of ganglion cells), segmental aganglionosis that does not involve the terminal rectum, and various forms of neuronal dysmorphogenesis in which ganglion cells are present, but dysplastic cytologically or deranged architecturally, should not be regarded as HSCR, even though the clinical presentation may be similar. It is very likely that the pathogeneses of these conditions are distinct from HSCR.
7.2.2.2 Genetics
HSCR is a multigenic disorder with incomplete penetrance. Isolated aganglionosis is found in approximately 70 % of patients; 12 % are associated with karyotypic abnormalities and 18 % of patients are cytogenetically normal but have additional anomalies (Chakravarti and Lyonnet 2001). The overwhelming majority of patients with isolated (nonsyndromic) HSCR harbor a mutation of the RET gene located on chromosome 10q11. RET encodes a tyrosine kinase that functions as a coreceptor for a variety of neurotrophic factors, including glial cell line-derived neurotrophic factor (GDNF). A wide variety of specific RET mutations have been observed, the most frequent of which is a single nucleotide polymorphism (designated rs2435357 or RET + 3C → T) located in a noncoding regulatory region that normally functions as a gene expression enhancer (Emison et al. 2005, 2011). The common feature of pathogenic RET mutations is they reduce net signaling activity through the receptor complex (Chakravarti and Lyonnet 2001). RET, which is expressed by enteric neural crest cells as they colonize the gut, influences many cellular properties (e.g., survival, proliferation, chemotaxis) that are required for normal enteric nervous system development.
In contrast with the RET + 3C → T polymorphism, mutations that affect exons or splice sites in the RET gene are less common and are detected in 15–20 % of patients with isolated HSCR. However, in patients with long-segment HSCR and/or a family history of HSCR, the incidence of such mutations approaches 50 % (Chakravarti and Lyonnet 2001; Amiel et al. 2008). A discrete subset of missense mutations in RET are responsible for the autosomal dominant syndromes, familial medullary thyroid carcinoma (FMTC), and multiple endocrine neoplasia-2A (MEN2A) and multiple endocrine neoplasia-2B (MEN2B) (Kapur 2005). Some patients (1–5 % in various series) with HSCR carry a RETMEN2A/FMTC allele and will develop endocrine neoplasia (Kapur 2005; Amiel et al. 2008), which has led to the recommendation that all HSCR patients be screened for MEN2A/FMTC mutations (Moore and Zaahl 2008).
HSCR is an incompletely penetrant phenotype in most families that transmit RET mutations, particularly those that affect noncoding parts of the gene like the RET + 3C → T polymorphism. Combinatorial interactions between mutant RET alleles and/or between a single-mutant RET allele and other genetic loci have been shown to influence penetrance of aganglionosis (Bolk Gabriel et al. 2002; Carrasquillo et al. 2002; Arnold et al. 2009). Environmental factors may also modify phenotypic expression; although with the possible exception of exogenous retinoids (Fu et al. 2010), specific teratogenic agents have not been identified.
HSCR is also a frequent component of many syndromes. Some of the syndromes and their genetic bases are listed in Table 7.5. Again, incomplete penetrance of HSCR in many of these syndromes likely reflects modifying influences of genetic background or environmental exposures, the specifics of which are incompletely understood. For example, the reported incidence of HSCR in Down syndrome is 2–15 % (Moore 2008). For some syndromes, including Down syndrome, the RET + 3C → T polymorphism influences penetrance significantly (Table 7.6) (de Pontual et al. 2007; Arnold et al. 2009).
Table 7.5
Selected genetic syndromes associated with Hirschsprung disease
Syndrome | Features | Inheritance | Gene (locus) |
|---|---|---|---|
Bardet-Biedl (includes McKusick-Kaufman) | Pigmentary retinopathy, obesity, hypogenitalism, postaxial polydactyly, renal anomalies, mild mental retardation | AR | ≥14 genes |
Cartilage-hair hypoplasia | Metaphyseal chondrodysplasia, sparse light-colored hair, immunodeficiency, cancer predisposition | AR | RMRP (9p13.3) |
Down | Mental retardation, hypotonia, abnormal facies, hypothyroidism, duodenal stenosis, cardiac malformation | Sporb | Trisomy 21 |
Goldberg-Shprintzen | Microcephaly, abnormal facies, mental retardation, coloboma, polymicrogyria | AR | KIAA1279 (10q22.1) |
Haddada | Central hypoventilation, autonomic defects, +/− neuroblastoma | AD | PHOX2B (4p12) |
Mowat-Wilsona | Microcephaly, abnormal facies, mental retardation, hypospadias, renal anomalies, agenesis of corpus callosum | AR | ZHFX1B (2p22) |
Multiple endocrine neoplasia type 2A | Medullary thyroid carcinoma, pheochromocytoma, parathyroid hyperplasia | AD | RET (10q11.2) |
PCWHa | Peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome type 4 | AD | SOX10 (22q13) |
Smith-Lemli-Opitz | Microcephaly, 2–3 pedal syndactyly, holoprosencephaly, hypospadias, mental retardation, growth retardation | AR | DHCR7 (11q13.4) |
Waardenburg type 4 (Waardenburg-Shah, ABCD assn.)a | Deafness and piebaldism (patchy hypopigmentation) | AD or AR | SOX10 (22q13) |
EDNRB (13q22) | |||
EDN3 (20q13) | |||
Pitt-Hopkins | Epileptic encephalopathy, dysmorphic facies, hyperventilation, dysautonomia | AD | TCF4 (18q21) |
X-linked aqueductal stenosis (HSAS/MASA association) | Hydrocephalus, aqueductal stenosis, adducted thumbs, absent corpus callosum, mental retardation | XL-R | L1CAM (Xq28) |
Table 7.6
Influence of RET + 3C > T polymorphism on penetrance of Hirschsprung disease in various genetic syndromes
Penetrance of HSCR (%) | Frequency of T allelea (%) | |
|---|---|---|
Nonsyndromic HSCR | 0.02 | 80 |
RET-dependent syndromes | ||
Down | 5 | 45 |
Bardet-Biedl | 10 | 60 |
Haddad | 20 | 40 |
RET-independent syndromes | ||
Mowat-Wilson | 60 | 25 |
Waardenburg-Shah | >80 | 18 |
General population | 0 | 25 |
Mutational analysis may be helpful in the context of syndromic HSCR to confirm a specific genetic disorder, counsel families regarding recurrence risks and complications that may arise in other organ systems, or perform prenatal diagnostic testing. However, the clinical value of mutational analysis in nonsyndromic HSCR is debatable. With the exception of focused studies to exclude RET MEN2A/FMTC mutations, neither more comprehensive molecular genetic analysis of RET nor analysis of other genes is likely to affect management or genetic counseling significantly. Insufficient data exists to determine whether specific genetic alterations alter prognosis (e.g., postoperative complications). Incomplete penetrance and the combinatorial effects of HSCR susceptibility loci prohibit definitive prenatal diagnosis based on the presence/absence of a given mutation. At present, the best estimates for recurrent HSCR in the sibling or child of an affected family member are empirical based on the gender of the patient, family history, and length of the aganglionic segment (Table 7.7) (Chakravarti and Lyonnet 2001; Amiel et al. 2008).
Table 7.7
Empirical recurrence risks of nonsyndromic Hirschsprung disease
Proband’s gender | Length of aganglionic segment | Likelihood of recurrence (%) | |||
|---|---|---|---|---|---|
Sibling | Offspring | ||||
Female | Male | Female | Male | ||
Male | Rectosigmoid | 1 | 4 | ~0 | ~0 |
Descending colon | 7 | 10 | 9 | 11 | |
Proximal to splenic flexure | 8 | 11 | 13 | 18 | |
Female | Rectosigmoid | 2 | 6 | ~0 | ~0 |
Descending colon | 10 | 13 | 11 | 15 | |
Proximal to splenic flexure | 18 | 23 | 22 | 28 | |
7.2.2.3 Clinical Manifestations
HSCR is usually diagnosed in the first 6 months of life due to obstructive symptoms (failure to pass meconium, abdominal distension, emesis). Failure to pass meconium in the first 48 h after delivery is a common sign observed in 50–90 % of affected neonates (Klein et al. 1984; Martin and Torres 1985). Less frequent presentations include diarrhea or intestinal perforation. However, 10–15 % of cases are identified after a year of life and adult presentations occur infrequently (Crocker and Messmer 1991; Wu et al. 1995). In retrospect, many of the latter patients have a long history of obstipation and enema use (Blisard and Kleinman 1986).
7.2.2.4 Radiographic Features
Supine and left-lateral decubitus films show gaseous distension of bowel loops with fluid levels (Blake 2000). The pattern may suggest large intestinal distension with a transition to non-dilated distal bowel, which is aganglionic. Barium enema more definitively documents the abrupt transition from narrow distal aganglionic to proximal ganglionic bowel and is considered an imperfect, but fairly reliable, diagnostic method. Problematic cases are usually long-segment disease, particularly total colonic aganglionosis. Anorectal manometry is another fairly reliable diagnostic procedure for HSCR (Holschneider 2000). Manometry detects changes in the myoelectrical properties and pressures of the internal anal sphincter, in response to rectal distension. The normal response of sphincter relaxation is lost in patients with HSCR, who lack inhibitory innervation of the sphincter smooth muscle.
7.2.2.5 Pathology
7.2.2.5.1 Submucosal Biopsies
Aganglionosis can be diagnosed by evaluation of either submucosal or myenteric ganglia. Since in either instance the pathologist must establish absence of ganglion cells, adequate sampling is essential and the recognition of ancillary histopathological findings that correlate with aganglionosis can be helpful. In most centers, initial diagnosis of HSCR is based on histopathological study of suction rectal biopsies (Qualman et al. 1999). Diagnostic biopsies must be taken at least 1–1.5 cm proximal to the anorectal squamocolumnar junction, measure >2 mm in diameter, and include sufficient submucosa (Aldridge and Campbell 1968; Weinberg 1970; Venugopal et al. 1981). Typically, about one-third of the cross-sectional area of a biopsy should be submucosal tissue (Fig. 7.2). Although a suboptimal biopsy may exclude the diagnosis if submucosal ganglion cells are present, biopsies that include squamous epithelium or too little submucosal tissue are inadequate to establish the diagnosis of HSCR (Fig. 7.2). Squamous epithelium indicates a biopsy too close to the anorectal junction, where physiological hypoganglionosis exists normally.
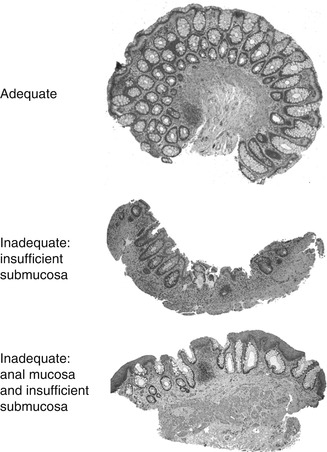
Fig. 7.2
Suction rectal biopsies to diagnose HSCR. An adequate biopsy is 2–3 mm long and one-third or more of the cross-sectional area is submucosa. Inadequate biopsies do not contain ganglion cells and either lack sufficient submucosa or contain anal mucosa (procured from the anorectal junction)
In principle, a single, adequate-size suction biopsy from the appropriate location is sufficient to exclude or diagnose Hirschsprung disease. Unfortunately, contemporary biopsy devices do not allow direct visualization of the anorectal canal and the location of a biopsy relative to the squamocolumnar junction is a rough estimate. However, the barrel of the biopsy device is scored so that the relative positions of multiple biopsies can be assessed. In order to reduce the likelihood of an inadequate sample and to facilitate the diagnosis of very short-segment disease, many centers advocate routine procurement of multiple biopsies at varying estimated distances from the anorectal junction (e.g., 1, 2, and 3 cm) (Qualman et al. 1999; Kapur 2009). In patients over a year of age, especially older children or adults, suction biopsy may be inadequate because the submucosa is thicker and more fibrous and submucosal ganglia are more widely spaced than in infants. Some surgeons opt for a deeper biopsy (e.g., incisional or forceps biopsy) in place of suction biopsy in toddlers and older patients.
When properly oriented and sectioned adequately (75–100 levels), H&E-stained, paraffin-embedded sections are generally sufficient to exclude the presence of submucosal ganglion cells and suggest the diagnosis of HSCR. Recognition of ganglion cells is generally easy, particularly with older patients, because the neuronal cell bodies have “mature” cytological features including abundant cytoplasm with basophilic Nissl substance. In neonates, especially premature infants, identification of ganglion cells is more difficult, because their “immature” cytology can be confused with endothelial cells, glia, or lymphocytes (Fig. 7.3). Nonetheless, experienced pathologists usually can discriminate even immature ganglion cells in H&E sections without any special techniques.
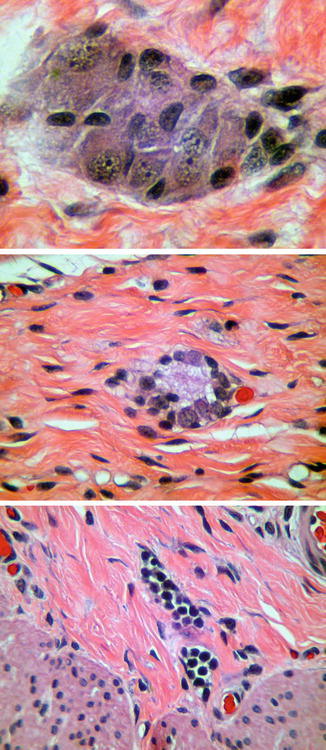
Fig. 7.3
Maturation of submucosal ganglion cells. Mature ganglion cells (top) are relatively easy to identify based on their size and staining characteristics. Immature ganglion cells (middle), as in the rectum of a neonate, have much less cytoplasm and are more difficult to distinguish from endothelial cells or lymphocytes (bottom)
The presence of hypertrophic nerve fibers is observed in many but not all cases of HSCR and helps establish the diagnosis (Monforte-Munoz et al. 1998). The hypertrophic nerves that exist in most patients with HSCR arise from extrinsic autonomic and sensory fibers, which enter along with vessels from the perirectal region and project for a finite distance rostrally (Meier-Ruge et al. 1972a; Mebis et al. 1990). It is the density and thicknesses of these fibers that increase in HSCR, giving rise to the “hypertrophic” nerves that are frequently, but not always, observed in the myenteric plexus, submucosa, and mucosa of HSCR patients (Fig. 7.4) (Howard and Garrett 1970; Tam and Boyd 1990; Wedel et al. 1999). In the rectal submucosa of a neonate or young infant, a nerve is considered hypertrophic when its thickness exceeds 40 μm (Monforte-Munoz et al. 1998). However, nerves as thick as 50 μm are present in toddlers and older children. Therefore, the patient’s age and density of hypertrophic nerves need to be considered, and the presence of a single thick submucosal nerve should not be regarded as diagnostic of aganglionosis. Because extrinsic nerves only project for a finite distance proximal to the rectum (Fig. 7.5), hypertrophic innervation may not be observed in biopsies taken rostrally in patients with long-segment disease (Fig. 7.6) (Meier-Ruge et al. 1972a). Furthermore, extrinsic nerve hypertrophy of the rectum may not be observed in patients with combined deficiency of intrinsic ganglion cells and other peripheral ganglia (probably more common with long-segment HSCR) or very premature infants with delayed extrinsic innervation.
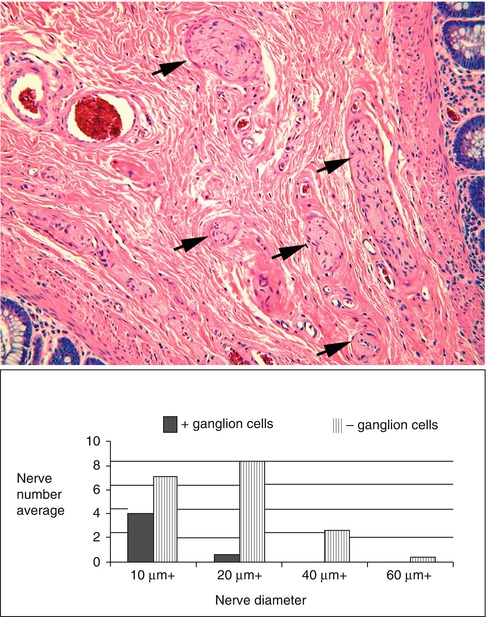
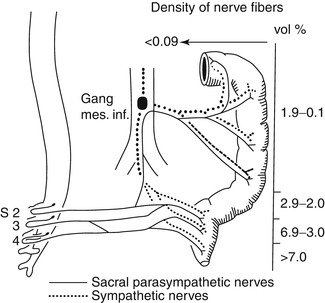
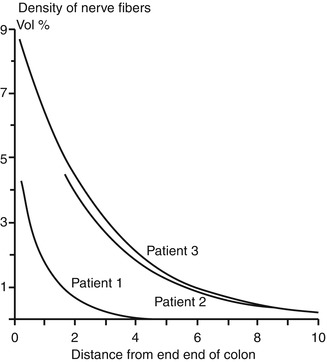
Fig. 7.4
Hypertrophic submucosal nerves in HSCR. Hypertrophic nerves (arrows) are present in the rectal submucosa in majority of patients with HSCR. Nerve diameters in excess of 20 μm (bottom) are rarely observed except in the context of congenital aganglionosis (Reprinted from Monforte-Munoz et al. (1998), with permission from Archives of Pathology & Laboratory Medicine. Copyright 1998. College of American Pathologists 122: 721–725)
Fig. 7.5
Sources of extrinsic innervation of the distal colon in HSCR. The primary sources of extrinsic innervation to the hindgut are the inferior mesenteric ganglion (sympathetic), sacral ganglia (parasympathetic), and sensory ganglia (not shown). As illustrated in this diagram (Based on data from seven patients with total colonic HSCR), the density of extrinsic innervation declines precipitously from the distal rectum to the splenic flexure (Reprinted from Meier-Ruge et al. (1972a). Copyright 1972, with permission from Elsevier)
Fig. 7.6
Variability in extrinsic innervation of the distal colon in HSCR. In some patients with total colonic aganglionosis (e.g., patient 1), hypertrophic extrinsic nerve fibers are confined to a very short length of the most distal rectum. Biopsies proximal to this area will be aganglionic, but will not contain hypertrophic nerve fibers (Reprinted from Meier-Ruge et al. (1972a). Copyright 1972, with permission from Elsevier)
The average submucosal biopsy is approximately 3 mm across and the average distance between submucosal ganglia varies with age, but is probably about 500 μm and contains 2–7 ganglion cells, each of which measures 20–30 μm in diameter. With 3–5 μm-thick paraffin sections, ganglia are missed in many random sections. Therefore, it is important to evaluate numerous sections (>75). With an adequate, well-oriented, not crushed, H&E-stained biopsy, one can be fairly confident about the diagnosis of HSCR simply by careful examination of each section. Absence of ganglion cells and the presence of multiple unequivocally hypertrophic nerves are diagnostic. In this situation, the presence of certain ancillary features of HSCR can be quite helpful, but is not required. The problem is that very often the biopsies are suboptimal due to paucity of submucosa, “crush” artifact, or both. For these cases, AChE histochemistry and calretinin immunohistochemistry are particularly valuable.
The frequent, but variable, presence of abnormally thick and numerous AChE-stained nerve fibers in the muscularis mucosa +/− lamina propria can be used to confirm the diagnosis, particularly when neither ganglion cells nor hypertrophic nerves are observed (Meier-Ruge et al. 1972b; Chow et al. 1977). The histochemical reaction used to visualize extrinsic nerve fibers only works with frozen sections of fresh or appropriately fixed tissues. Paraffin embedding inactivates the enzyme. Therefore, centers that perform AChE histochemistry regularly require at least two biopsies, one of which is frozen for AChE histochemistry and the other embedded in paraffin for H&E-stained sections.
The pattern of AChE staining in the rectum of normal children varies with age and includes staining of nerves in the submucosa and small fibers in the muscularis mucosa (Fig. 7.7). Usually, the latter are confined to the inner half of the muscularis mucosa. Some, but not all, of the rectal submucosal biopsies from HSCR patients contain more densely packed, large, AChE-positive fibers through the full thickness of the muscularis mucosa (Schofield et al. 1990). However, other abnormal patterns are also encountered, which sometimes are subtle and difficult to recognize (Pacheco and Bove 2008). In addition, prominent AChE-positive fibers may be present in the lamina propria. Lamina propria involvement should not be relied upon too heavily because hypertrophic nerves are restricted to the muscularis mucosa in biopsies from a significant subset of HSCR patients (Schofield et al. 1990).
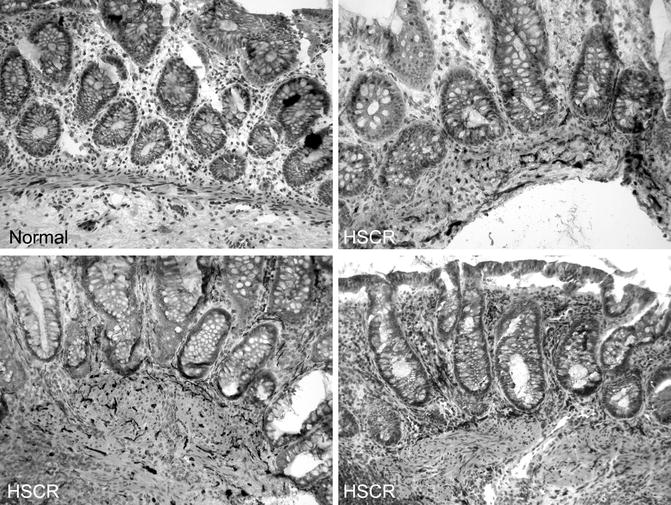
Fig. 7.7
AChE histochemical staining of frozen sections from submucosal biopsies. In biopsies from normal individuals, relatively thin, AChE-positive nerve fibers are confined to the deepest half of the muscularis mucosa and staining in the lamina propria is seldom seen. In contrast, darkly stained, thick fibers are present throughout the breadth of the muscularis mucosa in rectal biopsies from most HSCR patients. In addition, AChE-positive fibers are present in the lamina propria in some (lower two panels), but not all (upper right), biopsies from HSCR patients
Calretinin immunohistochemistry is an alternative ancillary diagnostic technique that is employed much like AChE histochemistry in the analysis of suction rectal biopsies. Calretinin is a calcium-binding protein that is expressed by a subset of submucosal and myenteric ganglion cells, some of which extend neurites into the mucosa (Furness 2006). In aganglionic suction rectal biopsies from an HSCR patient, calretinin-immunoreactive neurites in the lamina propria and muscularis mucosae are absent (Fig. 7.8) (Barshack et al. 2004). Be aware of the fact that calretinin-positive neurites may persist in the extrinsic nerves of the submucosa or myenteric plexus. The important diagnostic feature of aganglionosis is absent immunoreactive small nerves or individual neurites in the lamina propria and muscularis mucosae. In the context of appropriate positive and negative control sections, absent calretinin immunoreactivity has been shown to be equal, if not superior, to AChE histochemistry as a confirmative finding in HSCR (Guinard-Samuel et al. 2009; Kapur et al. 2009). Moreover, calretinin immunostaining can be performed on a paraffin section from a biopsy used for H&E analysis, thereby eliminating any need for additional biopsies or frozen sections.
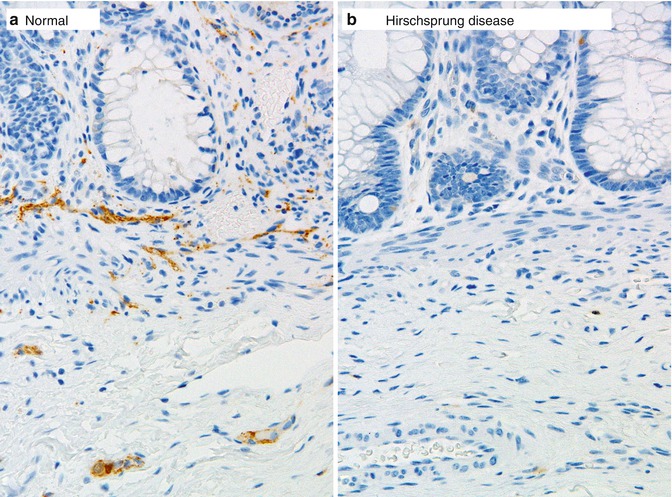
Fig. 7.8
Calretinin immunohistochemistry as an adjunct diagnostic method for HSCR. Immunoreactive nerve fibers are present in the muscularis mucosae and lamina propria of ganglionate bowel (a), but are completely absent in the mucosa of aganglionic bowel in Hirschsprung disease (b)
Considerable variation exists in the approaches taken by clinicians and pathologists to diagnosis of HSCR. In particular, the number of biopsies, number and thickness of sections, and use of AChE or calretinin staining differ between groups (Qualman et al. 1999). The approach used by the author is shown in Fig. 7.9 and incorporates calretinin immunohistochemistry as part of a routine procedure. AChE histochemistry can be substituted for calretinin immunohistochemistry but requires an additional frozen biopsy. The author believes that routine use of either ancillary method is advisable to ensure that technical and interpretative proficiency are maintained. However, others reserved AChE histochemistry or calretinin immunohistochemistry for cases when a biopsy is suboptimal (usually too little submucosa) and neither ganglion cells nor hypertrophic nerves are visualized in H&E-stained paraffin sections. Regardless, a strong HSCR-type AChE-staining pattern or complete lack of calretinin-immunoreactive nerves in the mucosa supports the diagnosis of HSCR. A normal or equivocal ancillary test result must be factored with clinical data (e.g., barium enema, family history, clinical severity), in order to decide whether to re-biopsy or manage conservatively. As with so many aspects of surgical pathology, the most important constant in HSCR diagnosis is clear communication between the clinician and pathologist, particularly about equivocal results.
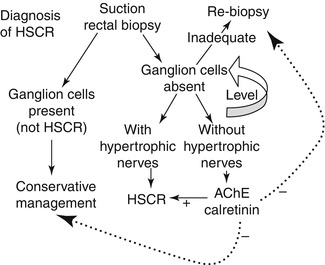
Fig. 7.9
Practical approach to evaluation of suction rectal biopsies for HSCR. If ganglion cells are present, then HSCR can be excluded. If ganglion cells are not present, the biopsy should be leveled to exhaust available submucosal tissue. If ganglion cells still are not identified, the pathologist must determine whether a biopsy is adequate based on its size, integrity, orientation, and the presence/absence of anal mucosa. If inadequate, repeat biopsy is needed. The presence of hypertrophic nerve fibers, even in a marginally adequate biopsy with no ganglion cells, is generally sufficient to diagnose HSCR. AChE or calretinin staining is most useful for aganglionic biopsies with limited submucosa or no hypertrophic nerves. A “positive” HSCR-staining pattern favors the diagnosis. A negative result must be integrated with clinical data to guide the next step in management of the patient, as indicated by the dashed lines
7.2.2.5.2 Intraoperative Seromuscular Biopsies
Many surgical approaches to HSCR are currently employed with a trend toward one-stage procedures that are transanal. In some instances, diagnosis based on suction rectal biopsy is followed by a two-stage procedure that begins with placement of an ostomy proximal to the aganglionic segment. Intraoperative seromuscular biopsies are important to determine that ganglion cells are present at the level where the ostomy or anastomosis will be placed. During such surgery, aganglionic gut may not be biopsied, a commitment to resection of at least the distal rectum having been decided based on the preoperative biopsy diagnosis. The transition to ganglionic bowel is located by analysis of seromuscular or full-thickness biopsies examined by intraoperative frozen sections. Generally, this is accomplished by sequential biopsies, from distal to proximal, until ganglion cells are identified.
Seromuscular biopsies should be a minimum of 5 mm in length and extend for a depth of 3–5 mm, so as to include the longitudinal and most of the circular layers of the muscularis propria. Proper orientation of the biopsy for frozen sections greatly facilitates sampling and identification of ganglion cells. The goal is to cut perpendicular to the serosal surface, thereby visualizing both the muscle layers and their interface in the histological sections. Ten sections are usually sufficient to confirm/exclude aganglionosis. Recognition of ganglion cells is generally not difficult, although inflammation sometimes obscures their cytological features (Fig. 7.10). For unknown reasons, in many instances, eosinophils predominate in infiltrates of the myenteric plexus, both in the aganglionic gut and more proximally (Fig. 7.11) (Lowichik and Weinberg 1997).
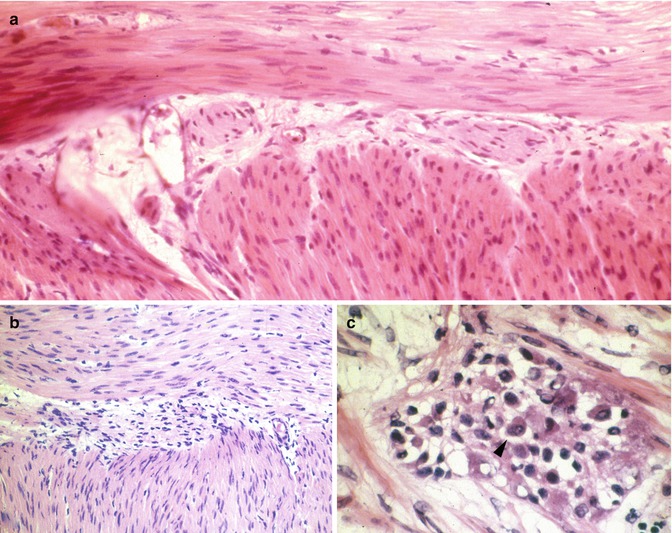
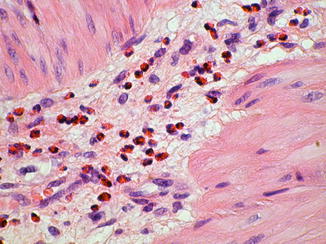
Fig. 7.10
Intraoperative seromuscular biopsies and HSCR. These images of the myenteric plexus from frozen sections illustrate absence (a, b) and presence of ganglion cells (c). In (a), hypertrophic extrinsic nerves are present in the myenteric plexus, but ganglion cell bodies are not. Inflammation, as illustrated in (b, c), complicates the diagnosis by obscuring the other cell types in the myenteric plexus. A ganglion cell is identified with an arrowhead
Fig. 7.11
Eosinophilic infiltrate in aganglionic myenteric plexus
Although perfectly controlled investigations have not been performed, the consensus from several published studies is that resection for HSCR should encompass the aganglionic bowel and any neuroanatomically abnormal transition zone, such that an ostomy or anastomosis is created with “normal” intestine (Langer 2004; Boman et al. 2007; Lawal et al. 2011; Coe et al. 2012). Intraoperative microscopic examination of a frozen section of the full circumference (“donut”) of the proximal resection margin is one way to reduce the likelihood of a transition zone pull-through. Features of transition zone to be excluded include aganglionosis or severe hypoganglionosis of large segments of the circumference and hypertrophic nerves in the submucosal or myenteric plexuses, as described below.
7.2.2.5.3 Analysis of HSCR Resections
The pathologist’s goals in analyzing resected aganglionic gut from an HSCR patient are to confirm the diagnosis of HSCR, establish the length of the aganglionic segment, and assess the integrity of the nervous system at the proximal end of the resection. Confirmation that the distal gut is aganglionic is straightforward (Fig. 7.12). If the proximal margin was examined intraoperatively by frozen sections, the findings should be confirmed by examining paraffin sections of the residual frozen. In addition, a permanent section of a second full-circumference tissue sample immediately adjacent to the proximal margin is also useful to confirm the frozen section findings with a well-oriented and well-fixed tissue that lacks any freezing artifacts. The approximate length of the aganglionic segment can be established by sampling a longitudinal strip of the resected bowel. Although not absolutely necessary, the author favors mapping the transition zone by sampling multiple areas along the length of the resected segment to document the presence/absence of submucosal and myenteric ganglia (Fig. 7.12). Because the interface between aganglionic and ganglionic gut is often irregular (Gherardi 1960; White and Langer 2000), it is prudent to map this area with closely spaced transverse sections, particularly if the transition is close to the proximal resection margin (Kapur 2009). Construction of a decent map may require repeated sampling sessions.
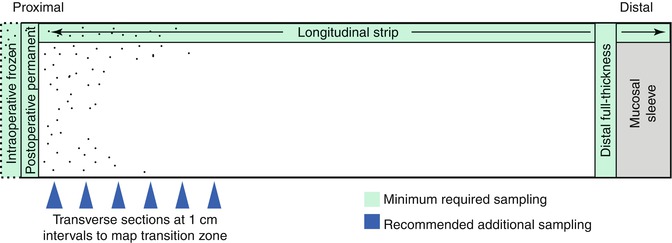
Fig. 7.12
Histological sampling of resected bowel from a patient with Hirschsprung disease. Transverse sections from the distal aganglionic bowel and proximal margin, including any residual tissue from intraoperative frozen section, should be submitted to confirm the diagnosis and exclude neuroanatomical pathology at the proximal margin (anastomosis site). A longitudinal strip of the entire length of the specimens can be used to crudely establish the length of the aganglionic segment at its proximity to the proximal margin. More detailed mapping of the transition zone can be accomplished with transverse sections at 1 cm intervals (blue arrows)
Examination of a full-circumference section from the proximal margin of the resection to exclude neuroanatomical defects is the most challenging and arguably the most important aspect of the process. In principle, the genetic etiologies that produce distal aganglionosis may have more subtle effects on the number, distribution, circuitry, and/or differentiation of proximal neurons (Amiel et al. 2008). Certainly, most HSCR patients harbor a transition zone of variable length (Fig. 7.13), in which the density of myenteric ganglia is obviously less than in normal gut (Gherardi 1960; White and Langer 2000; Boman et al. 2007). However, mild-to-moderate changes in neuronal density are extremely difficult to diagnose by routine analysis and yet may correlate with continued HSCR-like symptoms postoperatively (Swaminathan and Kapur 2010).
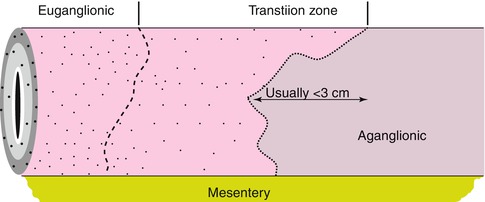
Fig. 7.13
Distribution of ganglion cells in the transition zone. The distal part of the transition zone is often characterized by partial circumferential aganglionosis, which typically extends less than 3 cm proximal to the completely aganglionic segment. The proximal transition zone is characterized by hypoganglionosis and hypertrophic nerves which can extend for highly variable distances. The proximal transition between hypoganglionic and euganglionic gut is gradual and cannot be resolved easily based on the density of ganglion cells in routine H&E-stained sections
In H&E-stained sections, the most frequently cited morphological features of transition zone are hypoganglionosis (oligoganglionosis) and hypertrophic nerves. In the distal transition zone, oligoganglionosis manifests as large portions of the circumference devoid of ganglion cells. Overlapping defects in the distribution of submucosal ganglion cells are common, but the correlation is not perfect. Regardless, priority is given to the distribution of myenteric ganglion cells because the normal distribution of submucosal ganglia is so sporadic and overt submucosal aganglionosis/hypoganglionosis seldom extends much farther rostrally than myenteric hypoganglionosis. In the proximal transition zone, a milder pattern of hypoganglionosis is characterized by a relatively normal circumferential distribution of ganglia, but small ganglia that consist of one or two ganglion cells with minimal neuropil (Fig. 7.14).
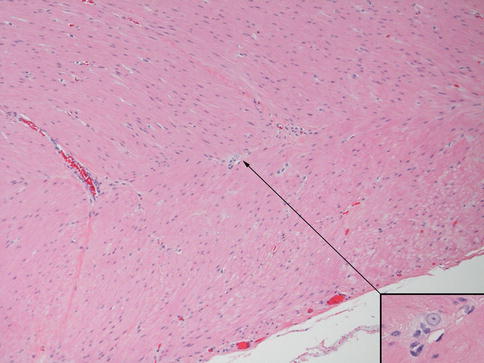
Fig. 7.14
Histopathological features of the transition zone: hypoganglionosis. Large portions of the circumference with widely spaced ganglia that each contains one or two ganglion cells and minimal neuropil are diagnostic of moderate-to-severe hypoganglionosis. Recognition of more subtle forms of hypoganglionosis requires formal neuronal counts with appropriate normative controls. Arrow indicates ganglion cell shown in inset
Hypertrophic submucosal and myenteric nerves, similar to those observed in aganglionic bowel, are another histopathological feature of the transition zone (Levitt et al. 2010). Such nerves can extend for several centimeters proximal to the aganglionic segment. These nerves share light and ultrastructural properties with extraenteric or serosal nerves, which arise from neuronal cell bodies outside the gut wall. Some, especially those in the submucosa, are thicker (>40 μm) than normal enteric nerves (Fig. 7.15). They appear more compact than normal intrinsic nerves of the enteric plexuses and, unlike the latter, are surrounded by perineurium. The perineural cells expressing Glut1 can be readily identified by immunostaining for this antigen (Kakita et al. 2000) (Fig. 7.15). The functional significance of hypertrophic nerves in ganglionic bowel has yet to be determined, but enlarged nerves are prevalent proximal to anastomoses in some patients with persistent postoperative obstructive symptoms (Coe et al. 2012). To avoid potential dysmotility associated with hypertrophic nerves, removal of transition zone with hypertrophic innervation has been recommended by at least one group (Lawal et al. 2011; Coe et al. 2012).
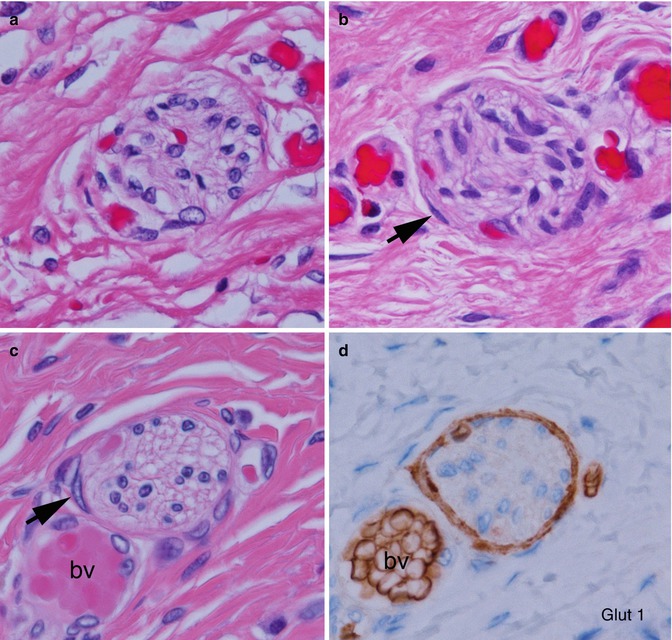
Fig. 7.15
Histopathological features of the transition zone: abnormal “extrinsic” nerves. (a) Normal submucosal nerve with inconspicuous perineurium and irregularly shaped and sized spaces in the neuropil. (b–d) Abnormal “extrinsic” submucosal nerves in the submucosa show more organized neuropil and glial cells and perineurium (arrows) that are Glut1-positive (d). Images (c, d) are adjacent sections and contain a blood vessel (bv) with Glut1-positive erythrocytes
In addition to hypoganglionosis, submucosal hyperganglionosis (intestinal neuronal dysplasia (IND) type B, discussed later in this chapter) has been reported in the proximal gut of HSCR patients (Puri et al. 1977; Meier-Ruge 1992; Moore et al. 1994). This entity is discussed more fully later in this chapter. Some investigators have gone as far as to suggest that serial biopsies of colon proximal to the aganglionic zone should be obtained to exclude IND before a pull-through procedure (Meyrat et al. 2001; Meyrat and Laurini 2003). However, insufficient compelling data exists to suggest that IND-like changes in HSCR either predict a poor outcome or should be managed any differently (Hanimann et al. 1992; Banani et al. 1996; Schmittenbecher et al. 1999; Haricharan et al. 2008; Coe et al. 2012).
7.2.2.5.4 Skip Lesions and Zonal Aganglionosis
It is important to be aware of two rare forms of congenital aganglionosis that deviate from the classic pattern, in which the distal rectum and uninterrupted contiguous bowel are devoid of nerve cell bodies (Kapur et al. 1995). In the intestinal tracts of persons with “segmental” (“zonal”) aganglionosis, ganglion cells are present in the distal rectum but are absent from a proximal segment of gut (Fig. 7.16). In contrast, “skip areas” are ganglion-cell-containing segments of large intestine, flanked proximally and distally by aganglionic gut (Fig. 7.16).
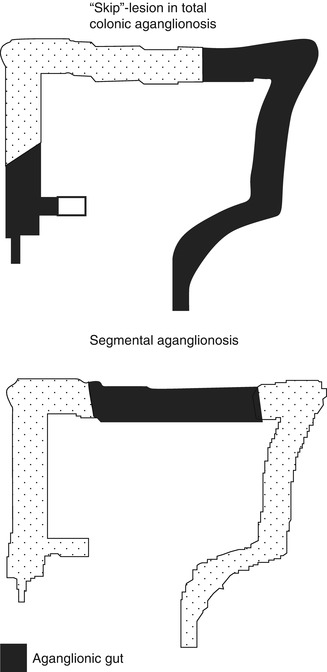
Fig. 7.16
“Skip”-lesions versus segmental aganglionosis
Segmental aganglionosis is considered to be an acquired lesion (disruption) that results when ganglion cells (or their precursors) in a fully colonized segment of gut die due to ischemic, viral, immunologic, or other types of injury (Yunis et al. 1983; Smith et al. 1997). The aganglionic segment can occur in small or large intestine. In some cases, a specific etiology is suggested by history (e.g., necrotizing enterocolitis), serological evidence for antineuronal antibodies, or other pathological findings (e.g., viral cytopathy). Alternatively, it has been suggested that segmental aganglionosis might result from failure of vagal and sacral crest cells to converge in the gut wall (Vaos 1989). At the time this hypothesis was introduced, it was less certain that sacral crest cells naturally adopt an enteric neural fate. Given recent evidence that a subset of colonic neurons derive normally from the sacral crest, the proposal is more tenable (Burns et al. 2000; Wang et al. 2011).
Skip areas are located in the large intestine and flanked by aganglionic areas that invariably include the distal rectum and appendix plus variable lengths of contiguous large and small intestine (Kapur et al. 1995). In a sense, a skip area is a ganglion-cell-containing segment that interrupts what otherwise would be an example of total colonic aganglionosis. A small skip area may involve only a small portion of the colonic circumference. In contrast, a large skip area can extend from the cecal region into the transverse or left colon. Pathologists and surgeons must be cognizant of skip areas and not use biopsies of the appendix as a means to diagnose total colonic aganglionosis since relatively large skip areas can be present in which ganglion cells exist. In at least one patient, the skip area was recognized, preserved, and used to establish a functional anastomosis between small intestine and anus (Martin et al. 1979).
7.2.2.5.5 Ultrashort-Segment HSCR
Ultrashort-segment HSCR is a controversial entity that has been defined differently by various investigators (Neilson and Yazbeck 1990; Ballard 1996; Meier-Ruge et al. 2004). Initially, the term “ultrashort-segment HSCR” was reserved to describe patients with clinical and radiological findings similar to those of HSCR, but with ganglion cells in their rectal biopsies (Davidson and Bauer 1958). The underlying assumption was that an extremely short aganglionic zone in the distal rectum, inferior to the submucosal biopsy site, was responsible for their symptoms. Others suggested that such patients had hypoganglionosis, which was unrecognized with contemporary pathological studies (reviewed by Ballard 1996), but convincing documentation of clinically significant distal hypoganglionosis is extremely difficult since ganglion cells are normally sparse or absent in the terminal 2–3 cm of the rectum (Fig. 7.17) (Aldridge and Campbell 1968).
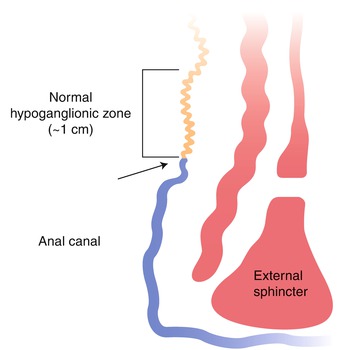
Fig. 7.17
Distribution of ganglion cells in the terminal rectum. A zone of hypoganglionosis exists in the distal 1 cm of the rectum, just proximal to the dentate line (arrow). In this zone, the density of submucosal ganglion cells and myenteric ganglion cells is reduced, and ganglion cells may not be present in a suction rectal biopsy
The author believes that “ultrashort-segment HSCR” should be reserved for patients with aganglionic distal rectal tissue and one or more additional histological feature of HSCR including abundant hypertrophic submucosal nerves and/or an unequivocal HSCR pattern of AChE histochemical or calretinin immunohistochemical staining (Kapur 2009). Other terms (e.g., internal sphincter achalasia) should be used for patients with clinical and manometric findings similar to HSCR, but with incomplete histological findings to support the latter diagnosis (Doodnath and Puri 2009). Conditions that mimic ultrashort-segment HSCR physiologically are likely to be heterogeneous given that regulation of anal sphincter tone is complex and is influenced by both the central and peripheral nervous systems (Stebbing 1998). Heightened sphincter tone could be caused by psychogenic, myogenic, or neurogenic etiologies. A major stimulus for sphincter relaxation is nitric oxide (NO) release by efferent nerve fibers in the smooth muscle (Buntzen et al. 1996). Reduced nicotinamide adenine dinucleotide phosphate (NADPH)-diaphorase activity (nitric oxide synthase), a marker for NO-producing nerve fibers, has been observed in the internal sphincter of patients with anal achalasia (Hirakawa et al. 1995; Moore et al. 1996). In some cases, the symptoms resolve after sphincter myotomy or Clostridium botulinum toxin injection (Krebs and Acuna 1994; Moore et al. 1996; Minkes and Langer 2000).
7.2.2.6 Treatment
HSCR is usually treated by surgical resection of the aganglionic bowel and anastomosis of proximal ganglionic bowel to the anus (Holschneider 2000). Transabdominal, laparoscopic, and transanal approaches and a variety of methods of rectal dissection/anastomoses have been developed. As avoiding damage to the internal anal sphincter and incontinence is a goal of every technique, a very short segment (~1–2 cm) of aganglionic distal rectum is retained and the distal margin of the resection may be at or just above the preoperative biopsy sites (Rabah 2010). The proximal margin should have a normal density and distribution of ganglion cells around the complete circumference of the bowel. In rare instances of small and large intestinal aganglionosis, parenteral nutrition and intestinal transplantation may be the only therapeutic options.
7.2.3 Congenital Hypoganglionosis
The term “hypoganglionosis” encompasses a broad range of enteric neuropathies that feature a reduced density of enteric ganglion cells. Hypoganglionosis can be congenital or acquired (Taguchi et al. 2006). The severe, and easiest to recognize, end of the hypoganglionosis spectrum are patients with a marked diminution of ganglion cells that is readily appreciated in H&E-stained histological sections without need for sophisticated morphometric analyses or special stains. More subtle variants of hypoganglionosis involve small or moderate reduction of neuronal density and/or loss of specific subtypes of enteric neurons, which can only be resolved by immunohistochemical studies and formal quantitative analysis with appropriate control data (Swaminathan and Kapur 2010; Knowles et al. 2011). In one published series, congenital hypoganglionosis accounted for 15 % of non-HSCR cases of intestinal neuronal malformation (Ure et al. 1997). However, severe hypoganglionosis is much less common. The following comments pertain to severe hypoganglionosis.
7.2.3.1 Etiology
The etiology of congenital hypoganglionosis is unknown. Despite some clinical, epidemiological, and pathological similarities to HSCR, genetic factors have not been shown to play a role in congenital hypoganglionosis, family history is typically unremarkable, and mutations have not been reported in HSCR susceptibility genes (e.g., RET) (Inoue et al. 2001).
7.2.3.2 Genetics
Given the variable penetrance and expressivity associated with established mutations that cause HSCR and the fact that the transition zone in HSCR is typically hypoganglionic, it seems reasonable to hypothesize that hypoganglionosis may result from identical genetic defects and pathogenic mechanisms as aganglionosis (Parisi and Kapur 2000). In fact, many proven or obligate carriers of HSCR gene mutations suffer from chronic constipation but have ganglion cells in biopsies of their rectal submucosa (Stannard et al. 1991). In addition, genetically engineered ret or gdnf mutations in mice produce severe hypoganglionosis (Shen et al. 2002; Carniti et al. 2006). However, a deliberate search failed to demonstrate either RET or GDNF mutations in a series of five patients with hypoganglionosis (Inoue et al. 2001) and neither familial recurrences nor specific genetic defects have been associated with hypoganglionosis to date.
7.2.3.3 Clinical Manifestations
Patients with congenital hypoganglionosis often present as neonates or young infants with signs of HSCR, but ganglion cells in their suction rectal biopsies (Dingemann and Puri 2010). Acute and chronic constipation, abdominal distension, and enterocolitis are all recognized clinical findings (Scharli and Sossai 1998). Males are affected three times more frequently than females, which is reminiscent of the gender bias observed in HSCR. In contrast with congenital hypoganglionosis, acquired hypoganglionosis can present at any age, depending to some extent on the underlying etiology. Neurodegenerative diseases that predispose to enteric ganglion cell loss are usually slowly progressive and most conditions that underlie acquired hypoganglionosis are more common in older children or adults. Chronic constipation or intestinal pseudo-obstruction is typical.
7.2.3.4 Radiological Features
The radiological features of congenital hypoganglionosis depend on the length of the hypoganglionic segment. In many cases, barium enema shows a narrowed length of distal hypoganglionic bowel preceded by a dilated segment of colon, similar to HSCR (Zhang et al. 2008). Contrast medium is retained proximal to the transition zone for an inordinately long period. In total colonic +/− small intestinal hypoganglionosis, dilated loops of gas-filled bowel may be present with either microcolon or no caliber change in the colon (Ikeda et al. 1988). Manometric studies usually demonstrate loss or insensitivity of the anorectal reflex.
7.2.3.5 Pathology
For reasons already discussed, subtle changes in the density or absolute number of enteric neurons are difficult diagnosis to establish with conventional histopathological studies of biopsies. Hence, hypoganglionosis is probably underdiagnosed except in severe cases (Krishnamurthy et al. 1993; Meier-Ruge et al. 1999). To avoid sampling bias, diagnosis requires either a segment of resected bowel several centimeters long or multiple full-thickness biopsies (Borchard et al. 1991). Suction rectal biopsies are insufficient because the distribution and sizes of submucosal ganglion cells are so variable (Watanabe et al. 1999). Moreover, hypoganglionosis may be limited to the myenteric plexus, so the density of submucosal ganglion cells is not considered relevant to the diagnosis. In H&E-stained sections, the pathology of isolated hypoganglionosis is identical to the transition zone of Hirschsprung disease; coexistent nerve hypertrophy is only present in the distal rectum. A reduced density of ganglion cell bodies is present in the myenteric and submucosal plexus. In severely affected bowel, normal ganglia are replaced by tiny ganglia composed of one or two ganglion cell bodies with little, if any, surrounding neuropil (Fig. 7.14). In young patients, ganglion cells appear smaller and less mature than appropriate. While they may mature as the patient ages, ganglion cell density does not improve.
Acetylcholinesterase histochemistry is regarded by some pathologists as an invaluable diagnostic study for diagnosis of HSCR (Dingemann and Puri 2010). In contrast with HSCR, hypoganglionosis has been associated with reduced AChE-positive innervation (Meier-Ruge and Bruder 2005). Subnormal AChE activity in a suction rectal biopsy that contains ganglion cells may warrant further investigation to exclude hypoganglionosis. However, clinical or pathological standards have not been established to determine when full-thickness biopsies should be obtained to exclude problems other than HSCR.
Although detailed anatomic studies of the entire gut are seldom reported, congenital hypoganglionosis almost always follows a pattern of intestinal involvement similar to HSCR, in that the distal rectum and a variable length of contiguous bowel are hypoganglionic (Scharli and Sossai 1998; Dingemann and Puri 2010). In addition, diffuse involvement of the entire colon and large parts of contiguous small intestine has been observed in infants with pseudo-obstruction (Ikeda et al. 1988; Kubota et al. 2001; Taguchi et al. 2006). Acquired hypoganglionosis shows greater variability with patchy zones of less severely affected bowel interspersed between near-aganglionic zones.
Another form of hypoganglionosis is selective absence of particular neuronal lineages from the enteric nervous system. In humans, the best evidence for this pattern of hypoganglionosis comes from silver-stained preparations of myenteric plexus (Fig. 7.1). Severe idiopathic constipation has been associated with selective loss of argyrophilic or argyrophobic neurons that was not apparent by routine H&E staining (Dyer et al. 1969; Krishnamurthy et al. 1985; Navarro et al. 1990). A large number of distinct neuronal subtypes have been identified in the gut wall based largely on different neurochemical profiles. Some of these subtypes correlate with particular functions (i.e., inhibitory vs. excitatory, motor vs. sensory vs. interneuron) (Furness 2006). However, silver-staining properties of enteric neurons have not been linked to a particular neurochemical, cytoarchitectural, or electrophysiological subtype of enteric ganglion cell.
A challenge for contemporary investigators is to develop practical diagnostic tests to assess normalcy of specific neuronal subtypes and to correlate changes in the latter with intestinal pseudo-obstruction. Some evidence that subtle changes in neurochemically distinct subsets of enteric neurons may underlie some disorders of intestinal motility comes from immunohistochemical analysis of 100 Australian children with slow-transit constipation (Wheatley et al. 1999). A reduced density of substance P (SP)-immunoreactive nerve fibers (excitatory innervation) has been observed in 30 % of patients (females more than males) with significantly slow colonic transit (Yik et al. 2011). Vasoactive intestinal peptide-immunoreactive fibers (inhibitory innervation) were reduced far fewer patients. Curiously, submucosal biopsies from a subset of the SP-deficient patients fulfilled diagnostic criteria for intestinal neuronal dysplasia type B (see below) (Imaji et al. 2000). Insufficient information is available to determine whether deficient SP nerve fibers correlate with reduced SP ganglion cells, but hypoganglionosis per se, is not a feature in these patients.
7.2.3.6 Treatment
Treatment recommendations for patients with congenital hypoganglionosis are similar to HSCR. Resection of the hypoganglionic gut may be curative. However, patients with extensive small intestinal involvement may require chronic parenteral nutrition or small bowel transplant. Early diagnosis and placement of a double-barrel jejunostomy have been suggested as a valuable management strategy in patients with such long-segment disease (Watanabe et al. 2011).
7.2.4 Acquired Hypoganglionosis (Degenerative Enteric Neuropathy)
Acquired aganglionosis is the consequence of ganglion cell death, which can be the outcome of intrinsic neurodegenerative disorders (e.g., neuronal nuclear inclusion disease) or extrinsic processes that destroy ganglion cells (e.g., Chagas disease). In principle, ganglion cell loss can be nonspecific or alternatively consist of degeneration of one or more specialized subset of enteric neurons. Examples of the latter phenomenon have yet to be well established as clinical entities, although disproportionate loss of neurochemically defined types of enteric neurons has been reported in patients with slow-transit constipation or other types of intestinal dysmotility (Taguchi et al. 1989; Hirakawa et al. 1995; Hutson et al. 1997; da Silveira et al. 2007a). Although some authors attach clinical significance to neuronal vacuolization and similar degenerative changes in enteric neurons without neuronal apoptosis or loss (Lindberg et al. 2009; Knowles et al. 2010), such histological changes should be interpreted very cautiously as similar alterations can be artifacts introduced by tissue handling.
7.2.4.1 Chagasic Enteropathy and Other Types of Postinfectious Hypoganglionosis
Loss of neurons and associated dysmotility coincides with, or is the sequel to, some viral or parasitic infections. Cytomegaloviral inclusions rarely occur in enteric ganglion cells including two patients with Hirschsprung disease (Dimmick and Bove 1984; Hershlag et al. 1984). However, no conclusive evidence exists that viral infection causes aganglionosis or any other type of dysmotility disorder. Associated lymphoplasmacytic ganglionitis was mild in these patients, and inclusions were present in other cell types in addition to ganglion cells. Herpes viruses would seem likely to produce enteric ganglionitis given that these viruses are neurotrophic, infect enteric neurons in experimental animals, and are associated with ganglionitis in other parts of the peripheral nervous system (Greenfield 1966; Gesser and Koo 1996). However, to the best of my knowledge, herpes viral enteric ganglionitis in humans has not been described. The best documented infectious cause of hypoganglionosis is Chagas’ disease.
7.2.4.1.1 Etiology
Chagas disease, the most widely recognized infectious etiology of pseudo-obstruction, is caused by the intracellular parasite Trypanosoma cruzi, which is endemic throughout much of Central and South America. The organism is carried in the alimentary tracts of several insect vectors and is transmitted via their fecal matter to many different mammals. The organism is also transmitted between humans congenitally or by blood transfusion. Infection is divided into acute and chronic phases (Koberle 1968). The acute phase occurs within weeks of initial infection and manifests in many patients as myocarditis and/or encephalitis. Acute Chagas disease usually has no gastrointestinal symptoms, although histopathology of the gut reveals inflammation in the smooth muscle and around and within ganglia. Organisms (amastigotes) may be visible in smooth muscle cells (Fig. 7.18) but are not usually present in ganglion cells.
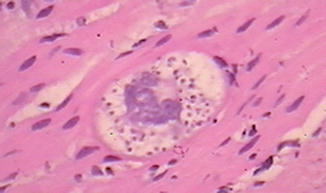
Fig. 7.18
Amastigote in the enteric smooth muscle of a patient with Chagas disease (Image kindly provided by Dr. Ricardo Drut)
Obstructive symptoms and distension of the gut in Chagas disease occur in regions with markedly deficient myenteric neurons and glial cells (Todd et al. 1969; da Silveira et al. 2007b). Ganglion cells appear to be lost primarily late in the acute phase and progressively during the chronic phase, during which time lymphocytic ganglionitis is present. Most patients do not develop pseudo-obstruction or megacolon until many years after the acute illness. Loss of ganglion cells is attributed to either an immune-mediated process or a neurotoxin, but not neuronal infection per se. With time, significant thickening of the muscularis propria due to smooth muscle cell hyperplasia occurs in the hypoganglionic gut. Eventually, these areas may be profoundly distended and virtually aperistaltic. In addition, a contributor to dysmotility may be autoantibodies that bind to and agonize muscarinic acetylcholine receptors expressed on enteric smooth muscle cells (Sterin-Borda et al. 2001).
7.2.4.1.2 Genetics
An estimated eight million persons in Central and South America and 300,000 person in the United States are infected with T. cruzi (Rassi et al. 2010). Over their lifetimes, 10–15 % of infected individuals will develop chronic digestive disease. Several studies suggest that variants of genes which encode tumor necrosis factors and other immunoregulatory molecules influence susceptibility to Chagas disease (Campelo et al. 2007; Alvarado Arnez et al. 2011). However, no predominant genetic risk factor has been identified.
7.2.4.1.3 Clinical Manifestations
Enteric dysmotility with megaesophagus and/or megarectum is a component of chronic disease. Megacolon frequently targets portions of the rectosigmoid and left colon and produces obstipation, abdominal distension, and occasionally obstruction.
7.2.4.1.4 Radiological Findings
Barium enema demonstrates marked dilatation of affected colon. Segmental distension of the sigmoid region is most frequent.
7.2.4.1.5 Pathology
Marked segmental colonic distension, smooth muscle hypertrophy, and hypoganglionosis are hallmarks of Chagasic megacolon. Organisms, which may be visible histologically in smooth muscle cells during acute infection (Fig. 7.18), are not present. However, a lymphocyte-rich infiltrate may surround or infiltrate myenteric and submucosal ganglia and nerves, including nerves that ramify through the muscularis propria (da Silveira et al. 2007b). The inflammatory cell population includes CD20-positive, CD3-positive, natural killer, and cytotoxic lymphocytes. Lymphocytic ganglionitis is present in patients with or without megacolon, but the proportion of natural killer and cytotoxic lymphocytes is significantly greater in distended colonic specimens. The densities of myenteric ganglion cells, glial cells, and peripherin-positive innervation are all greatly reduced with megacolon (Adad et al. 2001; da Silveira et al. 2007b).
7.2.4.1.6 Treatment
In early stages, medical management of Chagasic constipation is possible using laxatives or other agents. As the disease progresses, surgical resection of affected colon may be necessary. Antitrypanosomal agents are advised for patients with acute disease, but their value in patients with chronic disease is debated (Rassi et al. 2010).
7.2.4.2 Anthraquinone-Laxative Neurotoxicity
Several decades ago, Barbara Smith suggested chronic oral use of anthraquinone laxatives damaged the enteric nervous system and produced CIP (Smith 1968). She found reduced numbers of argyrophilic neurons and cytoarchitectural abnormalities in remaining argyrophilic neurons in the myenteric plexus of one patient and many mice exposed to these agents. Subsequent studies of other patients seemed to confirm her impression (Smith 1973; Oster et al. 1980; Riemann and Schmidt 1982). However, efforts by several different investigators to repeat Smith’s original findings in rodent models were generally unsuccessful (Dufour and Gendre 1984, 1988). Contamination of some anthraquinone preparations by unknown components, possibly non-glycoside-bound anthraquinones, was suggested as an explanation for the discrepancy based on apparent neurotoxicity of danthron, an unconjugated anthraquinone, when administered to rats. However, Heinicke and Kiernan conducted similar studies on rats using danthron and they found no evidence for neural loss (Heinicke and Kiernan 1990). In the latter studies, total neurons were evaluated in whole-mount preparations stained with cuprolinic blue and specific subtypes of neurons were evaluated immunohistochemically. Silver stains were not performed and it remains unclear whether laxative-induced changes in argyrophilia, rather than neuronal number, underlie the conflicting results in this body of literature.
7.2.4.3 Neuronal Intranuclear Inclusion Disease
7.2.4.3.1 Etiology
Neuronal intranuclear inclusion disease (NIID) is a hereditary disorder that affects the central and peripheral nervous systems. It is a neurodegenerative disorder characterized by progressive neuronal loss. Although the mechanisms responsible for cell loss are not known, analogies have been made to forms of spinal cerebellar ataxia and other hereditary neurodegenerative disorders in which intranuclear inclusions are found (Takahashi-Fujigasaki 2003). Many of the latter conditions result from expansions of trinucleotide (polyglutamine) repeat sequences in a specific gene. The gene product is one of several proteins that form an intranuclear inclusion, but it is unclear whether their accumulation has a toxic, protective, or bystander role in neuronal degeneration (Takahashi et al. 2000; Pountney et al. 2008).
7.2.4.3.2 Genetics
Despite the fact that most cases of NIID are sporadic, several well-documented familial examples suggest an autosomal dominant disorder due to mutations in an as yet unknown gene.
7.2.4.3.3 Clinical Manifestations
The clinical presentation of NIID is variable and may include any of a plethora of neurological symptoms including parkinsonism, cerebellar ataxia, chorea, dystonia, nystagmus, pyramidal tract signs, seizures, mental retardation, autonomic dysfunction, and peripheral neuropathy (Josephs 2011). Age of onset differs, but onset in childhood is most frequent. Intestinal dysmotility may be a minor component or may dominate the clinical picture (Schuffler et al. 1978; Barnett et al. 1992; El-Rifai et al. 2006).
7.2.4.3.4 Radiological Findings
Insufficient information exists to describe any characteristic radiological features of intestinal disease in these patients, although dilatation of proximal bowel (esophagus, stomach, and/or duodenum) has been the most consistent finding. “Nonpropulsive contractions of a dilated esophagus and small intestine” were observed in two adult siblings (Schuffler et al. 1978). The small bowel series from a neonate with NIID and pseudo-obstruction showed dilatation of the duodenum (El-Rifai et al. 2006).
7.2.4.3.5 Pathology
The characteristic histopathological feature is an eosinophilic circular intranuclear inclusion in myenteric and submucosal ganglion cells (Fig. 7.19). Such inclusions are often present simultaneously in neuronal nuclei in other parts of the peripheral nervous system, spinal cord, and brain, although functional deficits may not be evident in some of these sites (Erdohazi 1974; Malandrini et al. 1996; Goebel 1999). As the disease progresses, neuronal loss is typical and oligoganglionosis of both myenteric and submucosal plexuses may be severe.
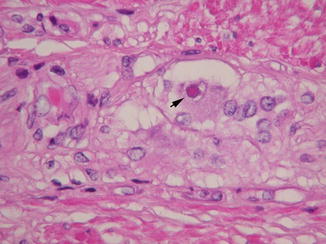
Fig. 7.19
Neuronal nuclear inclusion disease. A large intranuclear inclusion surrounded by a clear halo is present in a myenteric ganglion cell (arrow)
At least two families have been described in which intestinal pseudo-obstruction and intranuclear inclusions in enteric ganglion cells were prominent findings (Schuffler et al. 1978; Barnett et al. 1992). In each instance, the affected individuals were adults, but they exhibited signs and symptoms of pseudo-obstruction since childhood. Evidence for central nervous system involvement was clinically detected in all patients (e.g., ataxia, dementia, abnormal electroretinograms) and was confirmed pathologically in the brother and sister reported by Schuffler et al. (1978). The inclusions are generally larger (5–6 μm in maximal diameter) and more eosinophilic than neuronal nucleoli and surrounded by a clear halo (Fig. 7.19). Ultrastructurally, they are composed of tightly packed electron-dense filaments. They do not stain with Alcian Blue, Congo red, periodic acid-Schiff, Sudan black B, or methyl green. They are immunoreactive with antibodies specific for SUMO1, ubiquitin, and a variety of other nonspecific components (Pountney et al. 2008). Similar inclusions are found in autonomic and sensory ganglion cells, as well as in neurons and astrocytes of the brain and spinal cord. In the siblings reported by Schuffler et al., a third of all enteric ganglion cells contained inclusions and quantitative neuronal counts demonstrated a marked reduction in the density of ganglion cells throughout the gut. Analogous quantitative data was not presented for the family reported by Barnett et al. (1992). However, “a significant reduction of neurons” was specifically noted in biopsies from one patient.
Diagnosis of NIID from skin biopsies is possible (Sone et al. 2011). Nuclear inclusions with identical antigenic and electron microscopic features to those found in neurons are found in a subset of sweat gland cells, fibroblasts, and adipocytes.
7.2.4.3.6 Treatment
Treatment is symptom based and does not prevent progression.
7.2.4.4 Inflammatory Enteric Neuropathies
Generalized inflammation of the bowel wall, as occurs in Crohn’s disease, necrotizing enterocolitis, and a variety of other conditions, can also encompass the myenteric or submucosal plexuses and may account for some of the motility changes that accompany these conditions. Selective inflammation of ganglia and/or nerves is less common and usually associates with reduced intestinal motility. Eosinophilic inflammation of ganglia in the transition zone of Hirschsprung disease was discussed elsewhere in this chapter. Enteric ganglionitis occurs in three other contexts: infectious, paraneoplastic, and idiopathic. In each instance, T cells dominate the infiltrate and irreversible neuronal degeneration (“acquired aganglionosis”) is a potential outcome.
7.2.4.4.1 Paraneoplastic Ganglionitis
Intestinal pseudo-obstruction due to immune-mediated inflammation of myenteric +/− submucosal ganglia is a well-recognized complication of neoplasia (Lennon et al. 1991; Gerl et al. 1992; Chu et al. 1993; Condom et al. 1993; Anderson et al. 1996; De Giorgio et al. 2003). Neuroendocrine tumors including carcinoids and pulmonary small cell carcinoma appear particularly prone to associated enteric ganglionitis, and a 16-year-old girl was reported with ganglionitis and pseudo-obstruction that were diagnosed 1.5 years after a paravertebral ganglioneuroblastoma was successfully removed (Schobinger-Clement et al. 1999). The offensive tumor may be small and occult. Therefore, diagnosis of enteric ganglionitis necessitates a comprehensive search for neoplasia elsewhere.
Histologically, the inflammatory infiltrates are dominated by lymphocytes, predominantly T cells, plasma cells, and small numbers of histiocytes. Inflammation is usually confined to the ganglia and nerve plexuses but can affect any or all parts of the alimentary canal. The density of inflammatory cells is highly variable. Severe inflammation may obscure normal ganglionic cell types. Often, ganglion cells show degenerative features including hyperchromasia, nuclear pyknosis, or cellular dissolution. Non-enteric autonomic and sensory ganglia may be affected similarly. Even with successful treatment of the neoplasm, progressive neuronal loss and gliosis usually occur.
The pathogenesis of paraneoplastic ganglionitis is not conclusively established, but almost certainly has an immune basis. Reasonable titers of neuron-specific antibodies, particularly those that recognize nuclear antigens in CNS neurons (ANNA-1, anti-Hu), are generally present in the patient’s serum. These same antibodies bind to nuclear and/or cytoplasmic antigens expressed by enteric neurons. Presumably, lymphocytic infiltration of enteric ganglia results because of cross-reactivity with antigens produced by the neoplastic cells (De Giorgio et al. 2003). Immunosuppression may be helpful if it can be initiated before significant ganglion cell loss has occurred.
7.2.4.4.2 Idiopathic Ganglionitis
The clinical presentation and histopathological features of idiopathic ganglionitis are virtually identical to paraneoplastic ganglionitis, except no neoplasm is identifiable (Briellmann et al. 1996; De Giorgio et al. 2000). As in paraneoplastic ganglionitis, antineuronal antibodies can usually be documented in the patient’s serum and progressive neuronal loss to aganglionosis has been reported (Smith et al. 1997).
7.2.5 Diffuse Intestinal Ganglioneuromatosis and Associated Oncogenic Syndromes
A number of different patterns of increased enteric neuronal density have been reported, many in association with impaired peristaltic activity (Table 7.8). In virtually every instance, glial hyperplasia accompanies the added neurons. When neuroglial hyperplasia is relatively small and diffuse, it may be impossible to recognize in routine tissue sections, particularly when the general distribution of ganglion cells in myenteric and submucosal ganglia is preserved. However, moderate-to-severe ganglion cell hyperplasia frequently distorts the normal architecture of the enteric nervous system and surrounding tissues and is easier to identify. Rarely, neuroglial tumors assume immense proportions and mechanically obstruct the gut lumen. More often, impaired peristalsis relates to diffuse, multifocal, or segmental disruption of enteric neurophysiology caused either by the impact of excess ganglion cells within the neuromuscular circuitry or by underlying molecular alterations, which coincidentally promote hyperganglionosis. Multiple endocrine neoplasia type 2B (MEN2B), Cowden syndrome, and neurofibromatosis type 1 (NF1) are hereditary oncogenic syndromes in which diffuse ganglioneuromatous hyperplasia of the enteric nervous system and intestinal dysmotility often coexist (Carney et al. 1976; Ortonne et al. 1980; Chambonniere et al. 2003; Thway and Fisher 2009).
Table 7.8
Patterns of intestinal hyperganglionosis
Polypoid ganglioneuroma | Ganglioneuromatous polyposis | Diffuse ganglioneuromatosis | Intestinal neuronal dysplasia type B | |
|---|---|---|---|---|
Gross | Solitary polyp | Multiple polyps | Multiple polyps or diffuse thickening | Not visible |
Microscopic | Discrete mucosal +/− submucosal ganglioneuroma | Discrete mucosal +/− submucosal ganglioneuroma | Less discrete mucosal +/− submucosal +/− myenteric ganglioneuromas | ≥9 ganglion cells in ≥20 % of submucosal ganglia |
Motility symptoms | None | None | Constipation or pseudo-obstruction | Constipation or pseudo-obstruction |
Associated conditions | None | Juvenile polyposis, Cowden syndrome | MEN 2B, neurofibromatosis | None or variety of other primary obstructive lesions |
7.2.5.1 Etiology
Diffuse intestinal ganglioneuromas are hamartomas that form as a part of genetic disorders, which affect the proliferation of enteric neuroglial precursors and/or pattern formation in the gut wall. The kinetics of enteric ganglioneuroma growth are unknown, but progressive enlargement of existing lesions and the evolution of new lesions occur in many patients throughout life. A conditional PTEN-mutant mouse model is characterized by ganglioneuromatosis-like pathology, which results from abnormal proliferation of enteric neuroglial cells in the late fetal period and possibly postnatally (Puig et al. 2009).
7.2.5.2 Genetics
MEN2B, Cowden syndrome, and NF1 are autosomal dominant syndromes. MEN2B is cause by missense mutations in RET, most commonly a M918T substitution in the tyrosine kinase domain of the receptor, which alters its activity and substrate specificity (Ponder 2001; Raue and Frank-Raue 2010). Loss of function mutations in PTEN and NEUROFIBROMIN is responsible for Cowden syndrome and NF1, respectively (Farooq et al. 2010; Jett and Friedman 2010). Penetrance of intestinal ganglioneuromatosis is incomplete in Cowden syndrome and NF1 but is one of the defining clinical features of MEN2B. However, even among MEN2B patients, the severity of accompanying intestinal dysmotility is highly variable.
7.2.5.3 Clinical Manifestations
Diffuse intestinal ganglioneuromatosis may not produce any gastrointestinal symptoms or may be associated with constipation, diarrhea, and occasional chronic intestinal pseudo-obstruction. The majority of MEN2B patients suffer from impaired gastrointestinal motility, presumably as a consequence of their enteric neuropathy (O’Riordain et al. 1995). Dysmotility is less common in NF1 or Cowden syndrome, although gastrointestinal problems, including chronic intestinal pseudo-obstruction, occur in as many as 25 % of NF1 patients (Kim and Kim 1998). The pathogenesis of dysmotility is unclear; possible mechanisms include derangements of enteric neural physiology, physical constraints imposed by the hamartomas on contraction or relaxation of the bowel wall, and effects of hormones produced by the ganglioneuromas (Shulman et al. 1996). In rare instances, diarrhea has been attributed to production of vasoactive intestinal peptide by the hamartoma (Shulman et al. 1996; Moon et al. 2009).
Diffuse ganglioneuroma is benign process with no reported cases of malignant transformation. Despite the fact that neuroblastoma is a common pediatric tumor, primary enteric neuroblastoma is extremely rare (Ritter 1925; Barr et al. 1985; Kapur and Shepard 1992). The infrequency of intestinal neuroblastomas is particularly surprising given that enteric ganglion cells and glia share neural crest origin with sympathoadrenal derivatives, the sites of origin for most neuroblastomas. The relative differences in neuroblastoma development between the gut and elsewhere may reflect early divergence between enteric neural and sympathoadrenal lineages, which is evident in some experimental paradigms (Harris and Erickson 2007).
7.2.5.4 Radiological Findings
7.2.5.5 Pathology
Intestinal ganglioneuromas are composed of benign ganglion cells and associated glia (d’Amore et al. 1991; Shekitka and Sobin 1994). Although the lesions may show a patchy distribution along the length of the small and large intestines, colonic lesions have been most commonly reported as a source of symptoms (Thway and Fisher 2009). Diffuse ganglioneuromatosis refers to hyperplasia of neural and glial elements in both the submucosal and myenteric plexuses. The lesions are poorly demarcated, but may produce sessile or endophytic mucosal masses, segmental mural thickening, and vaguely nodular cut surfaces. Subtle lesions suggest simple enlargement of the normal enteric ganglioneuronal plexuses, which displace adjacent muscle or mucosal tissue while preserving some normal relationships (Fig. 7.20a). More dramatic examples consisting of nodular or multinodular masses of neuroglial tissue infiltrate the full thickness of the intestinal wall and/or project into the lumen. Cytologically, the neural and glial cells that compose these lesions resemble age-appropriate components of enteric ganglia, including ganglion cells with Nissl substance, eccentric nuclei with moderate-size nucleoli, and a background of enteric nerve-like neuropil. In children or adults, immature neuroblasts are generally not found, and mitotic figures are scarce. Neither immunohistochemistry nor electron microscopic studies are necessary to diagnose DIG, although such investigations indicate a variety of neurochemically distinct neuronal phenotypes in the hyperplastic process, similar to normal enteric ganglia.
Fig. 7.20
Ganglioneuromatous hyperplasia. (a) Ganglion cells and associated neuropil infiltrate the full thickness of the appendix wall in a child with MEN2B. (b) The mucosa of a polypoid ganglioneuroma contains abundant mature ganglion cells and associated neuropil. (c) The myenteric plexus is focally replaced by a plexiform neurofibroma in a patient with neurofibromatosis type 1
The widespread distribution and transmural infiltrative morphology of diffuse intestinal ganglioneuromatosis differ from the more focal or multifocal nature and relatively superficial location of solitary polypoid ganglioneuromas or ganglioneuromatous polyposis. The latter lesions are characterized by polypoid or sessile mucosa and/or submucosal lesions that form fairly discrete masses (Shekitka and Sobin 1994). Solitary polypoid ganglioneuromas are sessile or pedunculated lesions that usually measure less than two centimeters in diameter (Shekitka and Sobin 1994). Most are found in the colon and are not associated with any disturbance of intestinal motility. Two histological patterns have been described. The first is characterized by disrupted crypt architecture reminiscent of a juvenile polyp, but with collections of ganglion cell bodies and nerve fibers in the lamina propria (Fig. 7.20b). The second consists of a nodule that is comprised of abundant large nerves and ganglion cells in both the mucosa and submucosa.
Ganglioneuromatous polyposis is grossly similar to familial adenomatous polyposis in that numerous sessile or pedunculated lesions are found throughout the colon and occasionally in the distal ileum (Shekitka and Sobin 1994; Kanter et al. 2001). Intestinal dysmotility is usually not a problem. The lesions are histologically similar to polypoid ganglioneuromas involving mucosa, submucosa, or both. Ganglioneuromatous polyposis probably has an underlying genetic basis since many patients will have cutaneous or mesenteric lipomas. Furthermore, ganglioneuromatous polyps have been described in individuals and families with juvenile polyposis (Mendelsohn and Diamond 1984; Pham and Villanueva 1989), and ganglioneuromatous polyposis denotes cancer predisposition, as adenocarcinomas have arisen in some patients (Kanter et al. 2001). At present, it is unclear how ganglioneuromatous polyposis and juvenile polyposis are related, whether they share common genetic bases or represent phenotypic overlap between heterogeneous etiologies. Ganglioneuromatous polyps have been described in Cowden’s disease (Weary et al. 1972; Lashner et al. 1986).
While collections of heterotopic ganglion cells in the lamina propria are a shared feature of most forms of hyperganglionosis, it is important to realize that occasional non-clustered ganglion cells are found in normal gut mucosa, independent of any clinical dysmotility (Scurry 1990; Kramer et al. 2011). However, large groups of mucosal ganglion cells with or without associated nerves are abnormal, often correlate with intestinal symptoms, and may signify an underlying genetic disorder. This pathological pattern of heterotopic ganglion cells or grossly inconspicuous mucosal ganglioneuroma has been referred to as type 3 non-classifiable dysganglionosis (Berger et al. 2002).
In addition to intestinal ganglioneuromatosis, patients with neurofibromatosis are at risk for enteric neurofibromas that arise in the enteric nervous system, as in other parts of the peripheral nervous system (Bolande and Towler 1970; Hochberg et al. 1974; Feinstat et al. 1984). Glial cells and a fibromyxoid stroma predominate in neurofibromas in contrast to the enteric ganglion-like properties of a ganglioneuroma (Fig. 7.20c).
7.2.5.6 Treatment
Occasionally, dysmotility or obstruction caused by one or more bowel segment affected by intestinal ganglioneuromatosis can be so severe that surgical resection is required. If a specific oncogenic syndrome is identified, patients need to be monitored for development of associated neoplasms, families need to receive appropriate genetic counseling/screening, and prophylactic surgery (e.g., thyroidectomy for MEN2B) may be indicated. Somatostatin therapy may be effective in patients with secretory diarrhea due to vasoactive peptide secretion from ganglioneuromatous tissue.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


