Cystic Diseases of the Kidney: Introduction
Renal cystic disease comprises a wide range of disease entities. They can be classified as either (1) hereditary or acquired or (2) systemic or renal confined diseases that have the common feature of multiple renal cysts. Each disease entity differs in its presentation, prognosis, and management.
Renal cysts are smooth-walled, fluid-filled circular structures formed by focal outpouching of renal tubules. The pathogenesis of cyst formation has not been entirely elucidated. However, tremendous strides have been made in recent years. For autosomal dominant and autosomal recessive polycystic kidney diseases (ADPKD and ARPKD), a picture is starting to emerge. Defects in the primary ciliary sensing mechanisms, intracellular calcium regulation, and cellular cyclic AMP (cAMP) accumulation all seem to play a role in the altered cellular phenotype and functions.
Today, treatment includes risk modification, management of complications, and renal transplant or dialysis. There is no definitive therapy to eliminate or to retard cyst growth. A better understanding of its pathogenesis offers hope in the near future for correcting the underlying abnormalities in cystic pathways.
Autosomal Dominant Polycystic Kidney Disease
- Two renal cysts unilaterally or bilaterally before age 30 years by renal ultrasound in patients with a family history of ADPKD.
- Two cysts in each kidney between the ages of 30 and 59 years by renal ultrasound in patients with a family history of ADPKD.
- Four or more cysts in each kidney after age ≥60 years by renal ultrasound in patients with a family history of ADPKD.
ADPKD is the most common life-threatening monogenic disease. It occurs worldwide and in all races, affecting 1 in 400–1000 individuals. In the United States, approximately 500,000 people are affected and about 2000 begin hemodialysis each year.
ADPKD is an autosomal dominantly transmitted disease. It is composed of two types: ADPKD1, caused by mutations in the PKD1 gene and responsible for 85% of the clinical cases of ADPKD, and ADPKD2, caused by mutations in the PKD2 gene and accounting for approximately 15% of cases. A very small percentage of ADPKD patients, with a milder form of disease not linked to mutations in either PKD1 or PKD2, might have mutation(s) in a yet to be identified third PKD gene.
In addition to its renal manifestations, ADPKD is a multisystemic disorder with prominent extrarenal cystic and noncystic manifestations including polycystic liver disease and cysts in diverse organ systems (pancreas, arachnoid membrane, pineal gland, and seminal vesicles), intracranial saccular aneurysms, thoracic aortic aneurysms and dissections, coronary artery aneurysms, mitral and/or tricuspid valve prolapse, aortic valve insufficiency, aortic root dilation, and possibly colonic diverticula.
The severity of cystic renal dysfunctions is highly heterogeneous with significant interfamilial and intrafamilial variations. In general, ADPKD1 is more severe and is marked by an early onset of end-stage renal failure (mean age of 54 years) versus that of ADPKD2 (mean age of 74 years). Compared to female patients male ADPKD2 patients tend to develop more extensive cystic renal dysfunction, although they have less cystic liver disease.
With age, cystic renal enlargement occurs in all ADPKD patients. The severity of structural abnormality generally correlates with the renal manifestations including pain, hematuria, hypertension, and renal dysfunction. Massively enlarged kidneys can also cause compression of the neighboring organs and the inferior vena cava, leading to early satiety, dyspnea, and lower extremity edema.
Hypertension (HTN) occurs before the onset of renal failure in more than 80% of ADPKD patients. Although the prevalence increases with age, ADPKD is associated with an earlier onset and a significantly increased incidence of HTN compared to those in the general population.
The cause of HTN is multifactorial including activation of the intrarenal renin–angiotensin system, defects in nitric oxide endothelium-mediated vasorelaxation, elevated sympathetic activity, and possible defects in vascular smooth muscle cells directly associated with the PKD mutations.
Early onset and/or uncontrolled HTN are significant risk factors for faster renal disease progression and for mortality from cardiac complications such as left ventricular hypertrophy and coronary artery disease. Uncontrolled HTN can also worsen valvular heart disease and increase the risk of intracranial aneurysmal rupture and morbidity associated with rupture.
Episodes of acute flank pain are common. The potential etiologies are cyst hemorrhage, infection, stone, or, rarely, renal tumor. Massively enlarged kidneys can cause mechanical lower back pain. A small group of patients develops chronic flank pain without an identifiable etiology except for the enlarged kidneys. These patients are at risk for narcotic and/or analgesic dependence and medication-related complications.
Gross hematuria may be the initial presenting symptom. It occurs in up to 42% of ADPKD patients and can result from cyst hemorrhage, stone, infection, or renal tumor.
Most cyst hemorrhages are self-limited and resolve within 2–7 days. First episodes occurring in patients older than 50 years or episodes persisting for more than a week should be investigated to rule out neoplasm.
Occasionally, hemorrhagic cysts can rupture into the retroperitoneum causing retroperitoneal bleeding. This can be severe and life threatening.
A urine concentration defect, often associated with mild polyuria, is the most common and earliest manifestation of ADPKD. It usually goes unnoticed and is well compensated by adequate fluid intake.
Nephrolithiasis occurs in approximately 20% of ADPKD patients and is five times more common than in the general population. The majority of stones are composed of uric acid and/or calcium oxalate. Uric acid stones occur more frequently in ADPKD than in non-ADPKD stone formers. Factors that may contribute to the lithogenicity are urinary stasis due to distorted renal architecture, hypocitraturia, and low urinary pH (promoting uric acid stone formation). The symptoms and signs of nephrolithiasis are similar to those of non-ADPKD stone patients.
Whether urinary tract infections occur more frequently in ADPKD patients is unclear, but their risk of complicated infections is clearly increased. Infections include cystitis, pyelonephritis, renal cyst infection, and perinephric abscesses. They occur more frequently in females than in males. The main pathogens are Escherichia coli, Klebsiella, Proteus, and other Enterobacteriaceae. Symptoms and signs are urinary frequency and urgency for cystitis; fever, chills, nausea, vomiting, and flank pain for pyelonephritis, renal cyst infection, and perinephric abscesses.
ADPKD1 is associated with a 20-year earlier onset of end-stage renal failure compared to that of ADPKD2. Once the renal clearance starts to decline, it decreases linearly at a rate of approximately 5.0–6.4 mL/minute/year. Both genetic and environmental factors play a role in renal disease progression. Among patients with ADPKD1, the location of the PKD1 mutation may influence renal outcome. The mutations located in the first half of the PKD1 gene (5′region) were shown to be associated with a slightly earlier onset of renal failure compared to the mutations located in the second half of the gene (3′region).
Additional risk factors that portend a poor renal outcome include male gender, sickle cell trait, diagnosis of ADPKD before age 30 years, first episode of gross hematuria before age 30 years, hypertension before age 35 years, hyperlipidemia, low high-density lipoprotein (HDL), and cigarette smoking.
The symptoms and signs of renal failure in ADPKD, which begin to appear when the glomerular filtration rate is reduced to <30–40 mL/minute/1.73 m2, mirror those of non-ADPKD chronic renal failure.
Polycystic liver disease (PLD) is the most common extrarenal manifestation of ADPKD. Liver cysts originate from small clusters of intralobular bile ductules surrounded by fibrous tissue termed biliary microhamartomas and from peribiliary glands. The occurrence of PLD in ADPKD increases with age from 0% in ADPKD children to 20% in the third decade and over 75% in the seventh decade of life. Women, especially those with multiple pregnancies and/or on oral contraceptive or on estrogen replacement therapy, tend to have an earlier onset and worse PLD.
The majority of PLD patients are asymptomatic. When occurring, the symptoms usually result from either mass effect or cyst-related complications such as cyst hemorrhage, rupture, or infection. The liver synthetic functions are typically preserved because, despite even a significant degree of cystic liver involvement, the total amount of unaffected hepatic parenchyma is not reduced.
Symptoms associated with mass effect are dyspnea, orthopnea, early satiety, gastroesophageal reflux, mechanical back pain, uterine prolapse, rib fracture, and, in severe cases, failure to thrive. In rare cases, a massively enlarged cystic liver can cause obstructions to the hepatic venous outflow tract, portal vein and/or bile duct, or the inferior vena cava. These patients may develop portal hypertension, esophageal and/or gastric varices, ascites, and, rarely, obstructive jaundice.
Hepatic cyst hemorrhage and ruptures can present as acute abdominal pain, extrinsic bile duct compression, and liver enzyme elevation. Rarely, cysts can rupture into the peritoneum and cause acute ascites and life-threatening hemoperitoneum.
Patients with hepatic cyst infection may present with fever, chills, localized upper abdominal pain, leukocytosis, and elevation of alkaline phosphatase. Bacteremia is frequently present. The major pathogens are Enterobacteriaceae.
The incidence of intracranial aneurysms (ICAs) and ICA ruptures in ADPKD is increased by 5- to 10-fold compared to that in the general population. Family clustering is evident. Patients with a family history of ICA or subarachnoid hemorrhage (SAH, the consequence of ICA rupture) have an ICA occurrence of 21% versus 6% in those without such history.
The majority of ADPKD-associated ICAs are small (<7 mm in diameter) and are located in the anterior circulation (approximately 90%). Although compared to sporadic ones, ICAs in ADPKD have a younger mean age of aneurysmal rupture (39 versus 51 years).
The risk of ICA rupture (extrapolated from the International Study of Unruptured Intracranial Aneurysms) depends on the size and location of the ICAs and whether the patient has a prior episode(s) of SAH.
The yearly risk of rupture is less then 0.1% for the small sized (<7 mm in diameter) anterior circulation ICAs in patients without a prior SAH. The risk is higher for ICAs >7 mm in diameter or in the posterior circulation or in patients with a prior history of SAH.
Unruptured ICAs are generally asymptomatic. Rarely, patients can present with focal neurologic symptoms such as cranial nerve palsy or seizure due to local compression. Rupturing or ruptured ICAs typically present with prominent symptoms including episodes of sudden onset intense headache or headache with a quality different from that experienced before. The pain can radiate to the occipital and cervical region and may be accompanied by nuchal rigidity. Other associated symptoms are nausea, vomiting, photophobia, cranial nerve palsy, seizure, lethargy, and coma.
Other vascular manifestations more frequently seen (an approximately 10-fold increase) in ADPKD are thoracic aortic and cervicocephalic arterial dissections, intracranial arterial dolichoectasia, and coronary artery aneurysms. The symptoms and signs of these complications are similar to those seen in non-ADPKD patients.
Valvular heart diseases occur more frequently in ADPKD patients than in their nonaffected family members or the general population. Of those, mitral valve prolapse is the most common and can be detected by echocardiography in up to 20% of ADPKD patients. Other more frequent valvular heart diseases include mitral insufficiency, tricuspid insufficiency, tricuspid prolapse, and aortic insufficiency often associated with aortic root dilation. Symptoms vary from asymptomatic or episodic palpitations to, in rare cases, congestive heart failure. When a cardiac murmur is heard, antibiotic prophylaxis against subacute bacterial endocarditis is indicated.
Despite the frequent occurrence of hyperplasia and microscopic adenomas on renal pathology, the overall incidence of renal carcinoma in ADPKD is not increased. However, when occurring, the renal cancer tends to affect younger patients (mean age of 45 versus 55 years in the general population), be multifocal, and possess high-grade sarcomatoid features.
Grossly, numerous spherical cysts varying in size are obvious and are distributed evenly in both the cortex and medulla. The enlarged kidneys typically retain their reniform shape. However, renal tubular systems are distorted beyond recognition.
Microscopically, significant abnormalities are evident even in patients with mild renal insufficiency. These abnormalities include interstitial fibrosis frequently associated with inflammatory cell infiltration, tubular epithelial hyperplasia associated with flat nonpolypoid or polypoid hyperplasia and microscopic adenoma, and advanced sclerosis of preglomerular vessels including both afferent arterioles and interlobular arteries, which are more severe compared to those of non-ADPKD patients with the same degree of renal insufficiency.
Grossly, the size of cysts varies from pinpoint to huge. The cysts tend to cluster and spare segments of the hepatic parenchyma free of disease.
Microscopically, cyst walls are thin and lined with a single layer of flattened or cuboidal cells of biliary origin. Biliary microhamartomas are often seen in association with cysts on serial sections. Cysts derived from peribiliary glands can cause extrinsic compression of bile ducts.
Urinary abnormalities include reduction in maximal urine concentration, hypocitraturia and low urine pH, microscopic or macroscopic hematuria, and mild to moderate proteinuria.
Although renal ultrasonography (US) is efficacious for polycystic kidney disease (PKD) screening, it provides limited anatomic definition. Magnetic resonance imaging (MRI) and contrast-enhanced computer tomography (CT) can accurately assess renal volume, cyst volume, and preserved renal parenchyma. CT without intravenous contrast is sensitive in detecting and localizing renal stones (including uric acid stones) and hemorrhages (Figure 46–1). Both CT and MRI detect renal neoplasms with similar sensitivity. MRI is often preferred because nephrotoxic intravenous contrast can be avoided.
Figure 46–1.
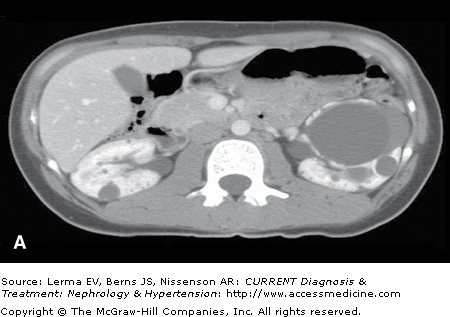
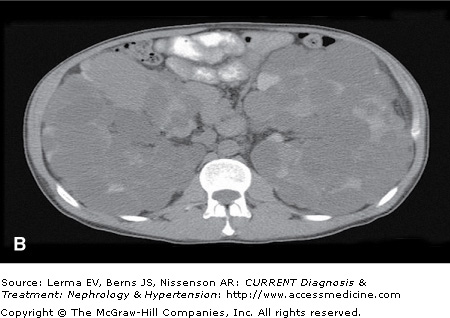
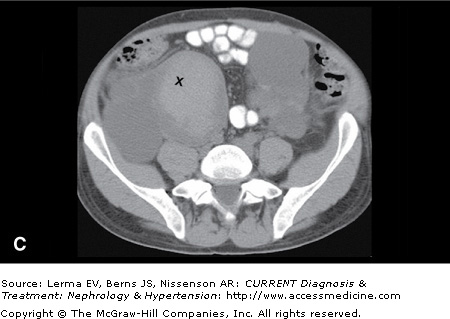
Autosomal dominant polycystic kidney disease. A: Contrast-enhanced CT scan in a 26-year-old female with early stage polycystic kidney disease. Note a dominant left renal cyst. B: Noncontrast CT scan in a 42-year-old male with advanced cystic disease and bilateral renal enlargement. Note innumerable renal cysts, many of which are hyperdense (hemorrhagic or with high protein content). C: The lower pole of the right kidney from the same patient in (B) contains a large cyst with increased and heterogeneous densities representing clots (marked by ‘X’) and recent hemorrhage.
To identify infected liver cysts, the combination of contrast-enhanced CT or MRI and nuclear scintigraphy may be helpful. CT or MRI might show thickened cyst walls, increased density of cyst contents, and air-fluid levels. Although limited by its imprecise anatomic definition, nuclear scintigraphy such as111 In-labeled leukocyte scans can help to localize the infected cysts. A definitive diagnosis lies in a positive identification of microorganism in cyst fluid by aspiration.
An MR or CT angiogram is the test of choice for detecting ICAs. It has an estimated sensitivity of >90% in detecting aneurysms ≥3 mm in diameters. Thin cut, noncontrast CT is the initial test of choice for identifying SAH. Hemorrhage appears as areas of increased density. If the CT result is equivocal and clinical suspicion is high, lumbar puncture should be performed to establish the diagnosis.
Renal cystic diseases can be a sequela of many systemic disease entities. This should be kept in mind especially when the patient’s overall presentation is atypical for ADPKD. A correct diagnosis lies in a careful identification of extrarenal manifestations that are not typically associated with ADPKD.
ADPKD must be differentiated from ARPKD, ACKD, multiple simple cysts, glomerulocystic kidney disease, tuberous sclerosis complex, and von Hippel–Lindau disease. The features separating ADPKD from these conditions are outlined in Table 46–1.
Disease | Inheritance | Extrarenal characteristics | Renal manifestations |
|---|---|---|---|
ADPKD | AD | Early adult onset of HTN; flank pain; gross hematuria; polycystic liver enlargement; SAH | Urine concentration defect; bilateral cystic renal enlargement; cysts arise from cortex and medullar; nephrolithiasis; renal failure with cystic disease progression |
ARPKD | AR | Infantile “Potter’s phenotype”; infantile HTN; portal hypertension: esophageal and gastric varices; hypersplenism | Large kidneys; bilateral fusiform collecting duct dilation; urine concentration defect; chronic renal failure |
ACKD | Not inherited | Longstanding renal failure; risk of renal cancer | Normal sized or small kidneys; cysts arise from cortex; cyst wall calcification, papillary cystadenomas and renal cancer |
Glomerulocystic kidney disease | AD (some) | Heterogeneous group of diseases | Cysts arise from the Bowman’s capsules |
Simple renal cysts | Not inherited | Associated with aging | Identified incidentally |
Tuberous sclerosis complex (TSC) | AD | Prominent skin lesions; CNS: Giant cell astrocytomas and cortical tubers; cardiac rhabdomyomas; pulmonary lymphangioleiomyomatosis | Cysts; renal angiomyolipomas; renal carcinomas (rare) |
von Hippel–Lindau disease (VHL) | AD | No skin lesion; retinal and CNS hemangioblastomas; pheochromocytomas; pancreatic cysts | Cysts; renal cell carcinomas (common) |
Orofacial syndrome type 1 | X-linked dominant | Cleft tongue and palate; broad nasal root; digital abnormalities | Cysts (may resemble ADPKD) |
Medullary cystic disease | AD | Hyperuricemia | Adult onset of renal failure |
Nephronophthisis | AR | Retinal degeneration (retinitis pigmentosa) | Childhood or adolescent onset of renal failure |
Medullary spongy disease | Usually not familial | Papillary calcifications and renal stones; normal glomerular filtration rate |
Currently, no definitive treatment modality is available to halt cystic disease progression or to induce cyst regression. Risk factor modification, early detection and management of ADPKD complications, and avoidance of potentially nephrotoxic agents are the mainstays of management aiming to limit morbidity and premature death.
In general, a diet of low sodium (<90 mEq/day), low cholesterol (<200 mg/day), high fiber, and 0.8 g/kg of ideal body weight/day protein is recommended. Avoiding or at least limiting caffeinated beverages to one to two servings per day is encouraged, because in vitro studies suggest that caffeine stimulates cAMP-mediated fluid secretion by the cyst-lining cells, which might promote cyst growth. Cessation of cigarette smoking should be stressed, because smoking is associated with a faster decline of renal function and an increased risk of ICA rupture.
Patients should be informed about and vigilant in monitoring the occurrence of ADPKD-associated manifestations because early detection and treatment can improve the overall disease outcome.
The best antihypertensive regimen for this patient population has not been clearly defined. In the majority of patients, angiotensin-converting enzyme inhibitors (ACEIs) and/or angiotensin receptor blockers (ARBs) are well tolerated and efficacious. If necessary, β- or combined α/β-blockers, a central α-adrenergic agonist, calcium channel blockers, or a low-dose thiazide diuretic (6.25–12.5 mg/day) may be added to optimize blood pressure control.
β-Blockers are indicated if patients have aortic root dilation or supraventricular tachycardia associated with mitral valve prolapse. High-dose diuretics should be avoided because they can reduce the renal blood flow and trigger acute gouty episodes.
Nonopioid agents are preferred for pain. Narcotic analgesics are reserved for managing the acute and severe episodes of pain exacerbation. Care should be taken to identify the underlying etiology and treat the correctable causes such as infection, renal stone, or tumor. Long-term use of potentially nephrotoxic analgesics such as combination analgesics or nonsteroidal anti-inflammatory drugs should be avoided.
Clinically significant chronic pain may be treated with tricyclic antidepressants after the correctable causes are ruled out and/or treated. Splanchnic nerve blockade with local anesthetics or steroids may provide prolonged pain relief in some cases and can be an adjunctive therapy.
Cyst decompression should be considered when conservative measures fail. Cyst aspiration followed by sclerosis and surgical or laparoscopic cyst decompression are the effective options.
For patients with a few dominant cysts deemed to cause pain, cyst aspiration followed by alcohol sclerosis (under US or CT guidance) can be carried out by an interventional radiologist and be both diagnostic and therapeutic. Minor complications include microscopic hematuria, localized pain, and transient fever. Severe complications, occurring mainly after aspirating the centrally located cysts, are rare including pneumothorax, perirenal hematoma, arteriovenous fistula, urinoma, and infections.
For patients with a large number of cysts contributing to pain, laparoscopic or surgical cyst fenestration can be effective for 80–90% at 1 year postoperation. Sixty to 80% have sustained pain relief for 2 years or longer. Laparoscopic or retroperitoneoscopic procedures have a shorter and less complicated recovery compared to open surgery. A relative contraindication is prior abdominal surgery with possible adhesions.
Other modalities that have been tried are laparoscopic renal denervation combined with cyst fenestration, laparoscopic or retroperitoneal nephrectomy, and embolization of the renal artery for patients with end-stage renal failure.
The majority of cyst hemorrhages are self-limited and respond to conservative management including bed rest, analgesics, and adequate fluid intake to prevent obstructing urinary blood clots. Rarely, the bleeding may be severe with formation of subcapsular or retroperitoneal hematoma, a fall in hemoglobin concentration, and, in severe cases, hemodynamic instability. These patients should be hospitalized for investigation by CT or angiography, volume resuscitation with transfusion if necessary, and, for refractory bleedings, arterial embolization or surgery.
Prompt treatment of symptomatic urethritis or cystitis with an oral antimicrobial is crucial in preventing retrograde seeding to renal parenchyma and/or cysts. Highly lipophilic antimicrobials such as trimethoprim-sulfamethoxazole, fluoroquinolones, or, rarely, chloramphenicol are the agents of choice. If available, the results of blood and urine cultures should also guide the selection of antimicrobial. Acute episodes of parenchymal or cyst infection usually require initial parenteral therapy.
If cyst infection persists after 1–2 weeks of treatment, percutaneous or surgical drainage should be considered. If fever recurs after the discontinuation of antibiotics, reevaluation is necessary to rule out complications such as obstruction, perinephritic abscess, or stone. If no complication can be identified, a prolonged treatment (several months) may be necessary to eradicate the infection.
The principles of stone management for ADPKD patients are the same as for idiopathic stone patients. Potassium citrate is a well-suited treatment for three main causes of stones in ADPKD: uric acid lithiasis, hypocitraturic calcium oxalate lithiasis, and distal acidification defects. For acute stone attack, treatment includes parenteral fluid, analgesics, and, when indicated, lithotripsy or urologic procedures.
ADPKD patients, in general, tolerate dialysis as well or better than patients with renal failure from other causes. This is possibly due to their higher production of endogenous erythropoietin and higher blood hemoglobin concentration. Rarely, hemodialysis can be complicated by episodes of intradialytic hypotension due to inferior vena cava compression caused by medially located right renal cysts or by hepatic cysts. This problem can be effectively managed by cyst aspiration or resection.
Peritoneal dialysis can also be safely carried out in ADPKD patients, although there is an increased risk of inguinal and umbilical hernias.
Transplantation is the treatment of choice for ADPKD end-stage renal failure. No difference is noted in posttransplant patient or graft survival between ADPKD and non-ADPKD patients. ADPKD-related complications such as mitral valve prolapse, aortic aneurysmal rupture, and hepatic or renal cyst infection are not adversely affected by renal transplant or posttransplant immune suppression.
Pretransplant cystic kidney nephrectomy is not routinely performed because native kidneys contribute to the maintenance of postoperative hemoglobin concentration and, in case of acute allograft failure, native kidneys may assist in fluid management. Pretransplant nephrectomy, however, is indicated if the patient has a history of cyst infection, frequent cyst hemorrhages, severe hypertension, or massive symptomatic renal enlargement.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


