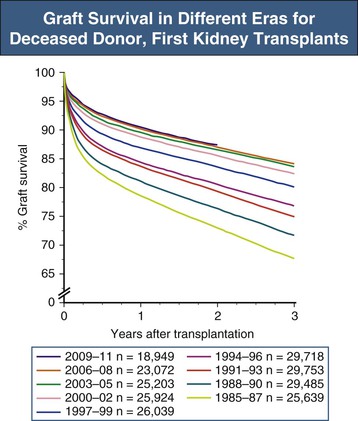
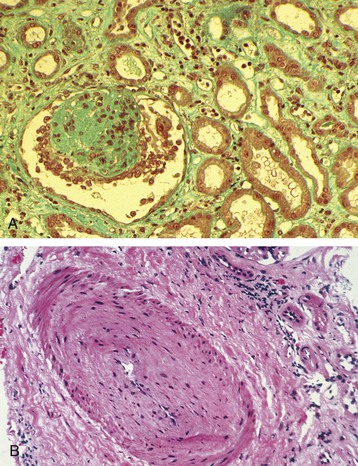
Christian Morath, Martin Zeier The results after kidney transplantation have steadily improved over the last 30 years, with a 1-year overall graft survival rate of about 94% and acute rejection rates within the first year after transplantation of 12%.1 Loss of an allograft because of acute rejection has become a rare event and predominantly occurs in patients who are presensitized against human leukocyte antigens (HLAs).2 Today, most allografts are lost because of a phenomenon called chronic allograft injury. Every year, about 4% of kidney grafts are lost (Fig. 107-1). Only a decade ago, it was believed that nonimmunologic causes were the main factors leading to chronic allograft injury and graft loss.3 However, our understanding has changed, and HLA alloantibodies are today believed to be the main culprit, accounting for more than half of graft losses in the long run.4 With the acknowledgment of alloantibodies as the major factor that contributes to graft failure, the term chronic allograft nephropathy has been replaced by more specific terminology in the Banff classification of renal allograft pathology (Box 107-1).5,6 A frequent pathologic feature of chronic allograft injury is interstitial fibrosis or tubular atrophy (see the later discussion of pathology and Fig. 107-2), but this is a nonspecific finding and does not imply any specific cause for the chronic allograft injury. Whereas immunologic causes are now believed to be mainly responsible for late allograft injury and graft failure, nonimmunologic causes still represent significant risk factors for (late) graft injury and graft loss. The donor graft itself may present with significant preexisting disease, limiting long-term graft survival.7 In addition, chronic interstitial fibrosis and tubular atrophy may result from early damage to the allograft such as ischemia-reperfusion injury or early acute rejection. Calcineurin inhibitor (CNI) nephrotoxicity, recurrent glomerular disease, and BK virus (BKV) infection but also chronic interstitial fibrosis and tubular atrophy originating from either hypertension or dyslipidemia further contribute to graft damage and graft loss in the long-run. Although some allografts are lost as a result of one specific cause, others accumulate the burden from several different causes with gradual loss of functioning nephrons. Main contributors to late graft injury and late graft loss are summarized in Box 107-2. Long-term graft survival is reduced in kidneys from older donors, an effect that is more pronounced in deceased compared with living kidney transplant recipients.8 Impaired graft survival is attributed to a differential response of the older organ to injury, an impaired capacity to withstand stress, a limited ability to repair structural damage, or amplification of external injury as a result of preexisting structural abnormalities.9 Reduced nephron mass resulting in glomerular hyperfiltration and hypertension with accelerated senescence is also a postulated mechanism for a progressive decline in graft function.10 The effect of kidney size mismatching is thought to be related to insufficient numbers of nephrons within the donor kidney, which in turn leads to compensatory hyperfiltration.11 Although there are experimental data to support this hypothesis, the extent to which an inadequate number of nephrons contributes to chronic injury in clinical transplantation is unknown. Female donor gender negatively affects kidney graft survival, with reduced survival when female grafts are transplanted into male recipients (risk ratio 1.22). This is not only a result of reduced nephron mass (“nephron underdosing”), because a comparable effect is found for cardiac allografts. Other mechanisms may include possible differences in the immunogenicity of male and female grafts.12,13 Graft survival is lower in recipients with longer ischemia times, and longer cold ischemia time represents a risk factor for the development of interstitial fibrosis and tubular atrophy at 6 months after transplantation.14 Ischemia-reperfusion injury may also trigger immune-mediated injury because ischemia and oxidative injury resulting from reperfusion are associated with upregulation of major histocompatibility complex (MHC) antigens, activation of the adaptive immune response, especially antigen-presenting cells and Toll-like receptors (TLRs), and release of proinflammatory cytokines, which can lead to acute rejection and subsequent interstitial fibrosis and tubular atrophy.15 Delayed graft function, commonly defined by the need for dialysis in the first week after transplantation, is a risk factor for interstitial fibrosis and tubular atrophy.16 There are also independent associations between delayed graft function and late graft failure and between delayed graft function and graft failure that is mediated by acute rejection. Major risk factors for delayed graft function are prolonged cold ischemia time, donor age older than 50 years, and the presence of HLA alloantibodies.2 BK virus is an endemic polyomavirus that is usually acquired during childhood and persists in the urinary tract. Although it is asymptomatic in the immunocompetent host, it becomes activated in the immunocompromised patient. Viremia may be found in 10% to 30% of patients within the first 6 months after transplantation, with biopsy-proven BKV-associated nephropathy in 1% to 10% of patients. BKV-associated nephropathy mimics the pattern of interstitial cellular rejection but may be distinguished in immunohistochemistry by a positive staining for the SV40 antigen. BKV-associated nephropathy, if untreated, may lead to severe interstitial fibrosis and tubular atrophy and graft loss in the majority of patients. The mainstay of therapy is the reduction of immunosuppression.17 The diagnosis and management of BKV nephropathy are discussed further in Chapter 105. Calcineurin inhibitor nephrotoxicity affects all histologic compartments of the transplanted kidney. Characteristic chronic CNI lesions include medial arteriolar hyalinosis, striped interstitial fibrosis, global glomerulosclerosis, and tubular microcalcification unrelated to other causes, such as tubular necrosis and hyperparathyroidism.18,19 CNI-induced arteriolopathy is characterized by nodular hyaline deposits in the media of afferent arterioles sufficient to cause narrowing of the vascular lumen.20,21 It is attributed to eosinophilic transformation and vacuolization of smooth muscle cells with subsequent necrosis. Arteriolar hyalinosis is the most reliable diagnostic marker of CNI nephrotoxicity. Confirmation of the diagnosis is made by exclusion of other causes, such as donor hyalinosis (which can be detected on the implantation biopsy specimen), diabetes, and hypertensive nephrosclerosis. Striped fibrosis is subjectively defined by a dense stripe of cortical fibrosis and atrophic tubules adjacent to normal cortex and is traditionally regarded as pathognomonic of CNI nephrotoxicity, but its appearance can be seen in any cause of fibrosis, especially if it is a result of microvascular injury. It is likely that the associated arteriolopathy and narrowing of the lumen contribute to development of fibrosis and atrophy after watershed infarcts within areas of ischemia. Local hypoxia leads to formation of free oxygen radicals, which promote cellular death by apoptosis. In addition, upregulation of transforming growth factor β (TGF-β) is considered an important etiologic factor in CNI toxicity.19 Calcineurin inhibitor toxicity was long believed to be the major factor contributing to late allograft loss. However, there has been a shift in this paradigm, and today CNI nephrotoxicity is believed to be responsible for only a smaller proportion of graft failures. The Long-Term Deterioration of Kidney Allograft Function (DeKAF) study demonstrated that 70% of patients with a primary or secondary histologic diagnosis of CNI nephrotoxicity had evidence of deposition of the complement split-product C4d and had donor-specific antibodies (DSAs) linking chronic graft failure to alloimmunity rather than CNI nephrotoxicity.22,23 Recurrent glomerulonephritis (GN) is diagnosed by exclusion of donor-transmitted disease and de novo GN. Its relative importance for graft loss increases as graft survival lengthens.24 Given that it is a relatively common occurrence and has implications for treatment and retransplantation, recurrent GN should be looked for carefully in patients with a prior diagnosis of GN. Diagnosis and management of recurrent disease are discussed further in Chapter 108. The vasculopathy in chronic allograft injury resembles systemic vascular disease, raising the possibility that conventional risk factors for cardiovascular disease may be implicated. Hypertension occurs in about 70% to 90% of transplant recipients. In a multicenter, retrospective study of 29,751 kidney transplant recipients, elevated systolic blood pressure above 180 mm Hg (compared with <140 mm Hg) at 1 year was associated with a doubled risk of graft failure at 7 years.25 Hypertension in these patients may be related to pretransplant hypertension, vascular disease, corticosteroid therapy, CNI therapy, or graft dysfunction. Dyslipidemias, including raised levels of total cholesterol, low-density lipoprotein cholesterol, and triglycerides, and cigarette smoking have also been associated with late graft failure. Some studies also suggest that hyperuricemia, which is common in patients with metabolic syndrome, may have a role in chronic allograft injury. The management of hypertension, dyslipidemia, and other cardiovascular risk factors in the transplant recipient is discussed further in Chapter 106.
Chronic Allograft Injury
Definitions and Epidemiology
Pathogenesis: Nonimmunologic Factors
Donor Age, Donor Gender, and Donor-Recipient Size Mismatching
Ischemia-Reperfusion Injury and Delayed Graft Function
BK Virus Nephropathy
Calcineurin Inhibitor Toxicity
Recurrent and De Novo Glomerular Diseases
Cardiovascular Risk Factors
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


