Claus G. Roehrborn, MD
Historically, voiding symptoms have been related to obstruction of the bladder outlet (Chapple et al, 2008). The traditional association in men is with the prostate, the so-called symptoms of “prostatism.” However, it is well recognized that voiding symptoms poorly correlate with underlying pathophysiology (de la Rosette et al, 1998). Similar symptoms can also be produced by any other form of obstruction, such as a urethral stricture or, conversely, by poor function of the lower urinary tract in circumstances in which there is impaired detrusor contractility. This has led to the recognition that, although LUTS commonly may be related to bladder outlet obstruction (BOO) as a result of benign prostatic obstruction (BPO), which is often associated with benign prostatic enlargement (BPE) resulting from the histologic condition of BPH, this is not invariably the case. For example, women also commonly present with voiding symptoms (Irwin et al, 2006). Failure to empty can be related to an outlet obstruction or to detrusor underactivity of the bladder or to a combination of both. Postmicturition symptoms, such as postvoid dribbling, occur in both sexes but most often in men, in whom these symptoms are highly common, are very troublesome, and cause significant interference with quality of life (Reynard et al, 1996). Storage symptoms are currently largely encompassed by the term overactive bladder (OAB) syndrome, which is defined as urgency, frequency, nocturia, and urgency incontinence and which is believed to be correlated with an underlying detrusor overactivity (Abrams et al, 2003). These symptoms tend to be more bothersome than voiding symptoms, especially if they are associated with incontinence. Storage symptoms in both sexes are commonly associated with urinary infections or, more rarely, with other conditions, such as bladder stones, carcinoma, or carcinoma in-situ in the bladder.
This understanding can be shown as partially overlapping populations (Fig. 91–1). Whereas many men older than the age of 40 will develop histologic hyperplasia (i.e., BPH), not all will have bothersome LUTS. Of those, some will and others will not develop measurable BPE. It is common for men to have BPE without having LUTS and vice versa. BOO may also be present with or without LUTS and with or without BPE; and in some cases BOO exists in men with BPH (e.g., from stricture) (Roehrborn, 2008).
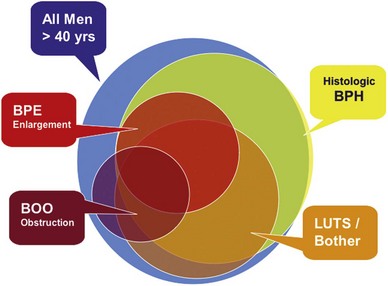
(From Roehrborn CG. Pathology of benign prostatic hyperplasia. Int J Impot Res 2008;20[Suppl. 3]:S11–8.)
Etiology
Histopathologically BPH is characterized by an increased number of epithelial and stromal cells in the periurethral area of the prostate and thus correctly referred to as hyperplasia and not hypertrophy, a term often found in the older literature. The observation of new epithelial gland formation is normally seen only in fetal development and gives rise to the concept of embryonic reawakening of the stroma cell’s inductive potential (Cunha et al, 1983; Isaacs, 2008). The precise molecular etiology of this hyperplastic process is uncertain. The observed increase in cell number may be due to epithelial and stromal proliferation or to impaired programmed cell death leading to cellular accumulation. Androgens, estrogens, stromal-epithelial interactions, growth factors, and neurotransmitters may play a role, either singly or in combination, in the etiology of the hyperplastic process.
Hyperplasia
In a given organ, the number of cells, and thus the volume of the organ, is dependent on the equilibrium between cell proliferation and cell death (Isaacs and Coffey, 1987). An organ can enlarge not only by an increase in cell proliferation but also by a decrease in cell death. Although androgens and growth factors stimulate cell proliferation in experimental models, the relative role of cell proliferation in human BPH is questioned because there is no clear evidence of an active proliferative process. Although it is possible that the early phases of BPH are associated with a rapid proliferation of cells, the established disease appears to be maintained in the presence of an equal or reduced rate of cell replication. Increased expression of antiapoptotic pathway genes (e.g., bcl2) supports this hypothesis (Kyprianou et al, 1996; Colombel et al, 1998) Androgens not only are required for normal cell proliferation and differentiation in the prostate but also actively inhibit cell death (Isaacs, 1984). In the dog, experimental BPH can be produced by androgens combined with estradiol (Walsh and Wilson, 1976; DeKlerk et al, 1979; Berry et al, 1986a; Juniewicz et al, 1994). Despite a significant increase in gland size there is actually a reduction in the rate of DNA synthesis compared with untreated controls (Barrack and Berry, 1987), indicating that androgens and estrogens both inhibit the rate of cell death. Neural signaling pathways, especially α-adrenergic pathways, may also play a role in balancing cell death and cell proliferation (Anglin et al, 2002).
The hyperplasia results in a remodeling of the normal prostatic architecture (Untergasser et al, 2005). Epithelial budding from preexisting ducts and the appearance of mesenchymal nodules characterize the early stages of the process, but the tissue phenotype of patients with established disease is highly variable.
BPH may be viewed as a stem cell disease (Barrack and Berry, 1987). Presumably, dormant stem cells in the normal prostate rarely divide, but when they do, they give rise to a second type of transiently proliferating cell capable of undergoing DNA synthesis and proliferation, thus maintaining the number of cells in the prostate. When the proliferating cells mature through a process of terminal differentiation they have a finite life span before undergoing programmed cell death. In this paradigm the aging process induces a block in this maturation process so that the progression to terminally differentiated cells is reduced, reducing the overall rate of cell death. Indirect evidence for this hypothesis comes from the observation that secretion, one parameter of epithelial cell differentiation, decreases with age, suggesting that the number of differentiated cells capable of secretory activity may be decreasing (Isaacs and Coffey, 1987). A survey of human BPH specimens for a marker of cellular senescence (senescence-associated β-galactosidase [SA-β-gal]) demonstrated a higher portion of senescent epithelial cells in men with large prostates, suggesting that an accumulation of those cells may play a role in the development of prostate enlargement (Choi et al, 2000). More recent studies support the hypothesis that impaired cell senescence may play a significant role in the etiology of BPH (Castro et al, 2003).
Hormones may exert their influence over the stem cell population not only with advancing age but also during embryonic and neonatal development (Naslund and Coffey, 1986). The size of the prostate may be defined by the absolute number of potential stem cells present in the gland, which in turn may be dictated at the time of embryonic development. Studies in animal models have suggested that early imprinting of prostatic tissue by postnatal androgen surges is critical to subsequent hormonally induced prostatic growth. As with the hormonal regulation of adult prostatic tissues, sex steroid hormones may exert their imprinting effect directly or indirectly through a complex series of signaling pathways (Lee and Peehl, 2004).
Role of Androgens
Although androgens do not cause BPH, the development of BPH requires the presence of testicular androgens during prostate development, puberty, and aging (McConnell, 1995; Marcelli and Cunningham, 1999). Patients castrated before puberty or who are affected by a variety of genetic diseases that impair androgen action or production do not develop BPH. It is also known that prostatic levels of dihydrotestosterone (DHT) as well as the androgen receptor (AR) remain high with aging despite the fact that peripheral levels of testosterone are decreasing. Moreover, androgen withdrawal leads to partial involution of established BPH (Peters and Walsh, 1987; Isaacs, 2008).
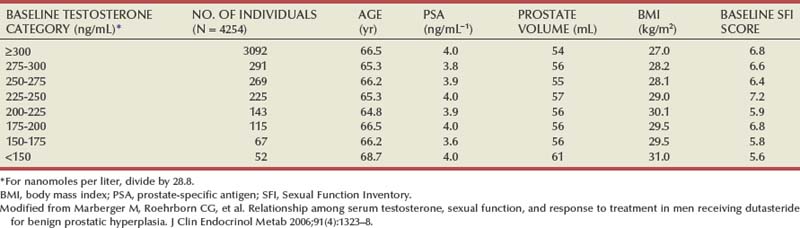
If normal ranges are assumed, there is no clear relationship between the concentration of circulating androgens and prostate size in aging men. In the Olmsted County Study of Urinary Symptoms and Health Status Among Men cohort (median age 60.9 years) serum bioavailable testosterone levels were found to decline with increasing age while the estradiol/bioavailable testosterone ratio increased (Roberts et al, 2004). Bioavailable testosterone correlated negatively and estradiol/bioavailable testosterone ratio positively with prostate volume, but this association was much less apparent after age adjustment. Baseline data from a large BPH medical therapy study confirmed the absence of a relationship between serum testosterone, serum prostate-specific antigen (PSA), and prostate volume (Marberger et al, 2006) (Table 91–1).
Table 91–1 Absence of Significant Relationship between Serum Testosterone and Serum PSA and Prostate Volume
In the brain, skeletal muscle, and seminiferous epithelium, testosterone directly stimulates androgen-dependent processes. In the prostate, however, the nuclear membrane bound enzyme steroid 5α-reductase converts the hormone testosterone into DHT, the principal androgen in this tissue (see Fig. 91–1) (McConnell, 1995). Ninety percent of total prostatic androgen is in the form of DHT, principally derived from testicular androgens. Adrenal androgens may constitute 10% of total prostatic androgen, although the importance of this stored hormone source in the etiology of BPH is negligible. Inside the cell, both testosterone and DHT bind to the same high-affinity androgen receptor protein (Chatterjee, 2003). DHT is a more potent androgen than testosterone because of its higher affinity for the AR. Moreover, the DHT-receptor complex may be more stable than the testosterone receptor complex. The hormone receptor then binds to specific DNA binding sites in the nucleus, which results in increased transcription of androgen-dependent genes and ultimately stimulation of protein synthesis (Andriole et al, 2004). Conversely, androgen withdrawal from androgen-sensitive tissue results in a decrease in protein synthesis and tissue involution. Besides inactivation of key androgen-dependent genes (e.g., PSA), androgen withdrawal leads to the activation of specific genes involved in programmed cell death (Kyprianou and Isaacs, 1989; Martikainen et al, 1990). Despite the importance of androgens in normal prostatic development and secretory physiology, there is no evidence that either testosterone or DHT serves as the direct mitogen for growth of the prostate in older men. Indeed, neither hormone is mitogenic to cultured prostatic epithelial cells (McKeehan et al, 1984). In the rat ventral prostate, differential gene expression experiments failed to demonstrate direct activation of mitogenic pathways (Wang et al, 1997). However, many growth factors and their receptors are regulated by androgens (see later). Thus the action of testosterone and DHT in the prostate is mediated indirectly through autocrine and paracrine pathways.
Androgen Receptors
The prostate, unlike other androgen-dependent organs, maintains its ability to respond to androgens throughout life. In the penis, AR expression decreases to negligible rates at the completion of puberty (Roehrborn et al, 1987; Takane et al, 1991a, 1991b). Thus, despite high circulating levels of androgen, the adult penis loses its ability for androgen-dependent growth. If the penis maintained high levels of AR throughout life, presumably the organ would grow until the time of death. In contrast, AR levels in the prostate remain high throughout aging (Barrack et al, 1983; Rennie et al, 1988). In fact, there is evidence to suggest that nuclear AR levels may be higher in hyperplastic tissue than in normal controls. Age-related increases in estrogen, as well as other factors, may increase AR expression in the aging prostate, leading to further growth (or to a decrease in cell death), despite decreasing levels of androgen in the peripheral circulation and “normal” levels of DHT in the prostate.
The potential role of AR mutations, polymorphisms, or other alterations in the pathogenesis of BPH is unclear (Chatterjee, 2003). A polymorphism in the number of CAG repeats (short vs. control) in the AR gene has been associated with larger prostate size and an increased risk of surgery (Giovannucci et al, 1999a, 1999b). However, another study from the Netherlands showed no relationship between the number of CAG repeats and BPH (Bousema et al, 2000). The later study also found no relationship between BPH and vitamin D receptor polymorphisms, although one Japanese study suggested an association (Habuchi et al, 2000). A more recent study of U.S. men showed a positive correlation between short CAG repeats and prostate volume, but a study of Finnish men found that short CAG repeats were significantly less common in men with BPH compared with control subjects (Mononen et al, 2002). Given the significant variation in reported findings, if short CAG repeats play a role in BPH pathogenesis it is likely to be minor (Hoke and McWilliams, 2008).
Dihydrotestosterone and Steroid 5α-Reductase
Intraprostatic DHT concentrations are maintained but not elevated in BPH. Initial studies of resected prostatic tissue suggested that prostatic DHT levels were higher in the hyperplastic gland than in normal control tissues. However, the controls used for these early studies were largely accident victims. Ongoing metabolism of DHT after death lowers the level of this androgen in cadaveric tissues. This was clearly shown in a study by Walsh and colleagues (1983) in which prostatic surgical specimens from men without BPH were used as the control. These investigators demonstrated that DHT levels are the same in hyperplastic glands as in normal glands. However, the aging prostate maintains a high level of DHT as well as a high level of AR; thus the mechanism for androgen-dependent cell growth is maintained. There is little question that androgens have at least a permissive role in the development of the disease process.
Two types of steroid 5α-reductase have been discovered, each encoded by a separate gene (Russell and Wilson, 1994). Type 1 5α-reductase, the predominant enzyme in extraprostatic tissues, such as skin and liver, is normally expressed in the 5α-reductase deficiency syndrome and is inhibited by dutasteride (Avodart) but not substantially by finasteride (Proscar). Type 2 5α-reductase is the predominant prostatic 5α-reductase, although it is also expressed in extraprostatic tissues. Mutations in the type 2 isoform are responsible for the clinical phenotype observed in the 5α-reductase deficiency syndrome. It is exquisitely sensitive to inhibition by finasteride and dutasteride (Carson and Rittmaster, 2003). Clearly, the type 2 isoform is critical to normal development of the prostate and hyperplastic growth later in life. The role of type 1 5α-reductase in normal and abnormal prostate growth remains to be defined. There is growing evidence to suggest that the type 1 isoform may play a more important role in prostate cancer compared with BPH, because increased levels of messenger RNA (mRNA) expression, protein, and functional enzymes have been demonstrated in prostate cancer (Thomas et al, 2008). Given that finasteride produces prostate size reduction identical to that with dual type 1/type 2 inhibitors and roughly equivalent to that with castration, it is unlikely that type 1–derived DHT is critical to hyperplastic growth.
Immunohistochemical studies with type 2 5α-reductase specific antibodies show primarily stromal cell localization of the enzyme (Thigpen et al, 1993; Silver et al, 1994). Epithelial cells uniformly lack type 2 protein, and some basal epithelial cells stain positively. Type 1 5α-reductase protein could not be detected in BPH or prostate cancer using initially available antibodies, although trace levels of type 1 mRNA have been seen in normal prostates, BPH, and cancer (Shirakawa et al, 2004). A study with a selective type 1 antibody demonstrated positive staining in only 7% of BPH cases (Thomas et al, 2003). In the same study, type 1 enzymatic activity was found in only 2 of 29 BPH specimens.
These data demonstrate that the stromal cell plays a central role in androgen-dependent prostatic growth and that type 2 5α-reductase within the stromal cell is the key androgenic amplification step. Thus a paracrine model for androgen action in the gland (Fig. 91–2) is evident. In addition, it is possible that circulating DHT produced in the skin and liver may act on prostate epithelial cells in a true endocrine fashion (McConnell, 1995). If dual type 1/type 2 5α-reductase inhibition has clinical utility over selective type 2 inhibitors, it is likely to be due to inhibition of peripherally produced DHT.
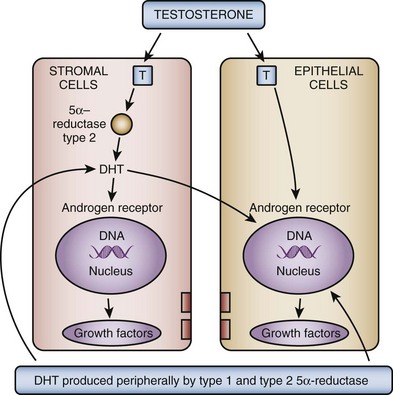
(From Roehrborn CG. Pathology of benign prostatic hyperplasia. Int J Impot Res 2008;20[Suppl. 3]:S11–8.)
Immunohistochemical studies in open enucleated BPH specimens show considerable intraprostatic and interprostatic 5α-reductase, making studies of its distribution based on single or one-time biopsy material very difficult (Sherwood et al, 2003).
Polymorphisms in type 2 5α-reductase (SRD5A2) have been reported, but their linkage to BPH is uncertain. The SRD5A2 gene on chromosome 2p23 frequently encompasses A49T and V89L substitutions and a TA dinucleotide repeat polymorphism. The 89L allele has been associated with lower enzyme activity, whereas the 49T allele has been associated with higher activity. Longer TA repeats are associated with mRNA instability and thus decreased enzyme activity. The number of L alleles, but not testosterone alleles or TA repeats, in one study correlated significantly with the presence of BPH (Salam et al, 2005). In the Olmsted County population, consistent associations between SRD5A2 genotypes and BPH were not demonstrated, although there was a weak correlation between V89L polymorphisms and prostate volume (Roberts et al, 2005).
Androgen withdrawal may partially exert its effect on the prostate through vascular effects (Buttyan et al, 2000). Castration induces acute and drastic vasoconstriction of blood vessels in the rat prostate (Hayek et al, 1999). This effect does not appear to be mediated through vascular endothelial growth factor (Burchardt et al, 2000). There is indirect evidence to suggest that abnormalities in the prostatic vascular system produced by other disease states (e.g., diabetes) may be a risk factor of BPH (Parsons et al, 2006; Parsons, 2007).
Role of Estrogens
There is animal model evidence to suggest that estrogens play a role in the pathogenesis of BPH; the role of estrogens in the development of human BPH, however, is less clear. In the dog, in which estrogens act synergistically with androgens to produce experimental BPH, estrogen appears to be involved in induction of the AR (Moore et al, 1979). Estrogen may, in fact, “sensitize” the aging dog prostate to the effects of androgen (Barrack and Berry, 1987). The canine prostate contains an abundance of high-affinity estrogen receptor (ER). In the dog, estrogen treatment stimulates the stroma, causing an increase in the total amount of collagen (Berry et al, 1986a, 1986b). There are at least two forms of ER. ER-α is expressed by prostate stromal cells, and ER-β is expressed by prostate epithelial cells (Prins et al, 1998). The estrogenic response of the prostate is determined by the type of ER present within the prostatic cells. Experiments in knockout mice suggest a “constraining influence” of estrogens on the prostate (Krege et al, 1998). In-vitro studies suggest that upregulation of ER-α in cultured prostate stromal cells is also associated with upregulation of fibroblast growth factor (FGF)-2, FGF-7, and other growth factors; the addition of androgens downregulated the ER and various stroma-derived growth factors (Smith et al, 2000, 2002).
Different actions may be mediated by the stromal ER-α and epithelial ER-β (Prins and Korach, 2008). Evidence also indicates that estrogen action mediated through the separate receptors may contribute to the etiology and progression of multiple prostate-diseased states (Table 91–2). These findings provide new avenues and alternative approaches for the treatment of prostate diseases including prostate cancer with novel therapies directed at ERs or estrogen metabolism. Because the two types of ERs may play distinct and perhaps opposing roles in many diseases of the prostate, including cancer progression, it is possible that receptor-specific agonists and antagonists may prove beneficial in therapeutic strategies in future clinical trials.
Table 91–2 Comparison of ER-α and ER-β Expression and Activities in the Prostate Gland
| ER-α | ER-β | |
|---|---|---|
| Localization | Stromal | Epithelial |
| Proliferation | Epithelial squamous metaplasia | Antiproliferative |
| Stromal proliferation | ||
| Differentiation | Epithelial dysplasia | Prodifferentiation |
| Immune response | Anti-inflammatory | |
| Antioxidant | ||
| Expression | Dysregulated in prostate cancer | Dysregulated in prostate cancer |
| Silenced in early cancers | ↓ Organ-confined disease | |
| Re-emergence with progression | ↑ In metastatic prostate cancer |
ER, estrogen receptor.
From Prins GS, Korach KS. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 2008;73(3):233–44.
Serum estrogen levels increase in men with age, absolutely or relative to testosterone levels. There is also suggestive evidence that intraprostatic levels of estrogen are increased in men with BPH. Patients with larger volumes of BPH tend to have higher levels of estradiol in the peripheral circulation (Partin et al, 1991). In the Olmsted County study cohort, in men with above median levels of bioavailable testosterone, the serum estradiol level correlated positively with prostate volume, even after adjusting for age (Roberts et al, 2004). Data on obesity, serum testosterone, estradiol, and prostate volume are conflicting (Zucchetto et al, 2005). Although there are relatively low concentrations of classic high-affinity ERs in human BPH (Farnsworth, 1996; Sciarra and Toscano, 2000), there may be a sufficient amount for biologic activity.
From experimental studies with aromatase inhibitors it appears that decreases in intraprostatic estrogen in animal models may lead to reduction in drug-induced stromal hyperplasia (Farnsworth, 1996, 1999). At present, however, the role of estrogens in human BPH is not as firmly established as the role of androgens. Species variation and cause-and-effect relationships are problematic.
Regulation of Programmed Cell Death
Programmed cell death (apoptosis) is a physiologic mechanism crucial to the maintenance of normal glandular homeostasis (Kerr and Searle, 1973). Cellular condensation and fragmentation precede phagocytosis and degradation, during which the apoptotic cell is phagocytosed by neighboring cells and degraded by lysosomal enzymes. Apoptosis occurs without activation of the immune system but requires both RNA and protein synthesis (Lee, 1981). In the rat prostate, active cell death occurs naturally in the proximal segment of the prostatic ductal system in the presence of normal concentrations of plasma testosterone (Lee et al, 1990). Androgens (presumably testosterone and DHT) appear to suppress programmed cell death elsewhere in the gland. After castration, active cell death is increased in the luminal epithelial population as well as in the distal region of each duct. Tenniswood (1992) suggested that there is regional control over androgen action and epithelial response, with androgens providing a modulating influence over the local production of growth regulatory factors that varies in different parts of the gland. Members of the transforming growth factor-β (TGF-β) family are likely candidates for this regulatory step (Martikainen et al, 1990).
In the rat prostate, at least 25 different genes are induced after castration (Montpetit et al, 1986). Normal glandular homeostasis requires a balance between growth inhibitors and mitogens, which respectively restrain or induce cell proliferation but also prevent or modulate cell death. Abnormal hyperplastic growth patterns, such as BPH, might be induced by local growth factor or growth factor receptor abnormalities, leading to increased proliferation or decreased levels of programmed cell death.
Stromal-Epithelial Interaction
There is abundant experimental evidence to demonstrate that prostatic stromal and epithelial cells maintain a sophisticated paracrine type of communication. The growth of canine prostate epithelium can be regulated by cellular interaction with the basement membrane and stromal cells. Isaacs and Coffey (1987), using a marker of canine prostatic epithelial cell function, demonstrated that epithelial cells grown on plastic quickly lose their ability to secrete this protein. In addition, the cells begin to grow rapidly and change their cytoskeletal staining pattern. In contrast, if the cells are grown on prostatic collagen they maintain their normal secretory capacity and cytoskeletal staining pattern and do not grow rapidly. This is strong evidence that one class of stromal cell excretory protein (i.e., extracellular matrix) partially regulates epithelial cell differentiation. Thus BPH may be due to a defect in a stromal component that normally inhibits cell proliferation, resulting in loss of a normal “braking” mechanism for proliferation. This abnormality could act in an autocrine fashion to lead to proliferation of stromal cells as well.
Further evidence of the importance of stromal-epithelial interactions in the prostate comes from the elegant developmental studies of Cunha, which demonstrate the importance of embryonic prostatic mesenchyme in dictating differentiation of the urogenital sinus epithelium (Cunha et al, 1980, 1983, 2003; Cunha and Donjacour, 1987; Cunha, 1994, 1996). The process of new gland formation in the hyperplastic prostate suggests a “reawakening” of embryonic processes in which the underlying prostatic stroma induces epithelial cell development (McNeal, 1990). Many of the prostatic stromal-epithelial interactions observed during normal development and in BPH may be mediated by soluble growth factors or by the extracellular matrix (ECM), which itself has growth factor–like properties. This model is even more intriguing, given the cellular localization of 5α-reductase (and thus DHT production) in the prostate stromal cell (Silver et al, 1994).
The complexity of the stromal/ECM/epithelial relationship is revealed in studies of the ECM signaling protein CYR61. CRY61 (an early immediate response gene) is an ECM-associated protein that promotes adhesion, migration, and proliferation of epithelial and stromal cells. A variety of growth factors increase the expression of CYR61 in both cell types, and the suppression of CYR61 expression by an antisense oligonucleotide significantly affects normal cell morphology (Sakamoto et al, 2003, 2004a, 2004b). CRY61 expression is significantly increased in human BPH tissues and is induced by lysophosphatidic acid (an endogenous lipid growth factor).
Growth Factors
Growth factors are small peptide molecules that stimulate, or in some cases inhibit, cell division and differentiation processes (Steiner, 1995; Lee and Peehl, 2004). Cells that respond to growth factors have on their surface receptors specific for that growth factor that in turn are linked to a variety of transmembrane and intracellular signaling mechanisms. Interactions between growth factors and steroid hormones may alter the balance of cell proliferation versus cell death to produce BPH (Fig. 91–3). Lawson’s group was the first to demonstrate that extracts of BPH stimulate cellular growth. This putative prostatic growth factor was subsequently found on sequence analysis to be basic fibroblastic growth factor (bFGF) (Story et al, 1989). Subsequently, a variety of growth factors have been characterized in normal, hyperplastic, and neoplastic prostatic tissue. In addition to bFGF (FGF-2), acidic FGF (FGF-1), Int-2 (FGF-3), keratinocyte growth factor (KGF, FGF-7), transforming growth factor-β (TGF-β), and epidermal growth factor (EGF) have been implicated in prostate growth. TGF-β is a potent inhibitor of proliferation in normal epithelial cells in a variety of tissues. In models of prostatic cancer there is evidence that malignant cells have escaped the growth inhibitory effect of TGF-β (McKeehan and Adams, 1988). Similar mechanisms may be operational in BPH (Salm et al, 2000), leading to the accumulation of epithelial cells (Kundu et al, 2008). Growth factors may also be important in modulating the phenotype of the prostate smooth muscle cell (Peehl and Sellers, 1998).
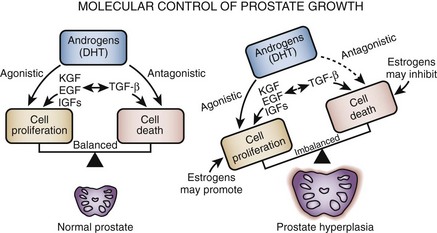
There is mounting evidence of interdependence between growth factors, growth factor receptors, and the steroid hormone milieu of the prostate (Rennie et al, 1988; Lee and Peehl, 2004). Although data on the absolute level of growth factor and growth factor receptors in hyperplastic as opposed to normal tissue conflict, it is likely that growth factors play some role in the pathogenesis of BPH. However, further research is necessary to establish the role of growth factors in a disease process in which cellular proliferation is not obvious.
If cellular proliferation is a component of the BPH process, it appears that growth stimulatory factors such as the FGF-1, FGF-2, FGF-7, and FGF-17 families, vascular endothelial growth factor (VEGF), and insulin-like growth factor (IGF) may play a role, with DHT augmenting or modulating the growth factor effects. In contrast, TGF-β, which is known to inhibit epithelial cell proliferation, may normally exert a restraining influence over epithelial proliferation that is lost or downregulated in BPH (Wilding et al, 1989; Sporn and Roberts, 1990, 1991; Peehl et al, 1995; Cohen et al, 2000; Lee and Peehl, 2004). TGF-β1 is a potent mitogen for fibroblasts and other mesenchymal cells but is also an important inhibitor of epithelial cell proliferation (Roberts and Sporn, 1993). TGF-β1 also regulates ECM synthesis and degradation and can induce cells to undergo apoptosis. In addition, TGF-β upregulates the production of bFGF-2, which is known to be an autocrine growth factor for prostate stromal cells (Story et al, 1993) and at least on one prostate smooth muscle cell line (PSMC1), TGF functions as an autocrine mitogen (Salm et al, 2000). Thus upregulation of TGF-β1 (which is expressed in prostate stromal cells) during BPH would favor expansion of the stromal compartment.
Indirect evidence to support this view comes from studies of reconstituted mouse prostate (Yang et al, 1997). Interestingly, the observation that TGF-β1 may regulate smooth muscle contractile protein expression suggests that TGF-β isoforms may be physiologic regulators of prostatic smooth muscle function (Orlandi et al, 1994). Cohen and colleagues (2000) found that stromal cells isolated from BPH specimens exhibited a blunted TGF-β growth inhibition relative to normal stromal cells and that the blunted response appeared to be due to a reduction in TGF-mediated increase in IGF binding protein 3 (IGFBP-3) expression. TGF-β may stimulate the overexpression of versican (chondroitin sulfate proteoglycan 2) in the ECM through inhibition of key metalloproteases (ADAMTS lineage) that normally degrade versican, leading to accumulation in the ECM (Cross et al, 2006). An increased risk for BPH was described in patients with a codon 10 polymorphism in TGF-β (Li et al, 2004).
The first evidence of increased FGF-2 levels in BPH came from the studies of Begun and coworkers (1995), who demonstrated a twofold to threefold elevation of FGF-2 in BPH compared with histologically normal glands. Further studies have demonstrated that both FGF-2 and FGF-7 are overexpressed in BPH tissues (Ropiquet et al, 1999). The major target of FGF-2 is thought to be the stroma itself (autocrine), although transgenic mice overexpressing FGF-2 develop glandular epithelial hyperplasia (Konno-Takahashi et al, 2004). KGF, a member of the FGF family (FGF-7), is produced in prostatic stromal cells (Yan et al, 1992). However, cell surface receptors for stroma-derived KGF are expressed exclusively in epithelial cells. As a result, FGF-7 (or a homolog) is the leading candidate for the factor mediating the stromal cell–based hormonal regulation of the prostatic epithelium. There is direct evidence that FGF-7 plays this role in the androgen-dependent mesenchymal-epithelial interactions involved in development of the seminal vesicle (Alarid et al, 1994). Abnormalities in stromal FGF-7 production or epithelial FGF-7 receptor could promote epithelial cell proliferation. Indirect evidence supporting this hypothesis comes from a study of transgenic mice overexpressing FGF-7 that develop atypical prostatic hyperplasia (Kitsberg and Leder, 1996). McKeehan’s laboratory demonstrated that FGF-10, a homolog of FGF-7, is expressed at high levels in the rat prostate, specifically in stromal cells of smooth muscle origin (Lu et al, 1999; Nakano et al, 1999). FGF-10 expression is increased by androgens and may have a mitogenic effect on prostate epithelium. Others studies suggest that cells expressing FGF-7 are localized in the stroma immediately adjacent to the epithelium, suggesting that the epithelial cells may induce FGF-7 expression. The paracrine factor most likely responsible for this effect is cytokine interleukin (IL)-1α (Giri and Ittmann, 2000; Lee and Peehl, 2004).
Some investigators have speculated that local hypoxia in the prostate (perhaps from atherosclerosis or other vascular events) is the initial event that induces FGF production (Lee and Peehl, 2004). Further growth of BPH nodules could impede blood flow, leading to further hypoxia (Parsons et al, 2008; Parsons and Kashefi, 2008). Hypoxia leads to upregulation of hypoxia inducible factor-1, which in turn increases the secretion of FGF-2 and FGF-7 from stromal cells.
Other growth factors implicated in BPH include FGF-17 (Polnaszek et al, 2004), FGF-10, and VEGF (Walsh et al, 2002). It remains difficult to ascertain which of the growth factors and growth factor receptors are key mediators of the BPH disease process and which are bystanders.
A unique animal model provides additional evidence that FGF-like factors may be involved in the etiology of BPH. A transgenic mouse line expressing the Int-2/FGF-3 growth factor demonstrated androgen-sensitive epithelial hyperplasia in the male mouse prostate histologically similar to human and canine BPH (Tutrone et al, 1993).
Insulin-like growth factors, binding proteins, and receptors also appear to be important modulators of prostatic growth, at least as it relates to cell growth in culture (Peehl et al, 1995; Lee and Peehl, 2004). A transgenic mouse model with overexpression of IGF-1 demonstrated prostate gland enlargement (Konno-Takahashi et al, 2003). Studies of BPH tissue demonstrate a higher concentration of IGF-2 in the periurethral area than in the peripheral zone (Monti et al, 2001). A study of Chinese men demonstrated a significant correlation between circulating IGF-1 and IGFBP-3 level and BPH (Dahle et al, 2002), but a study of the Olmsted County cohort failed to demonstrate any relationship between serum IGF-1 and prostate volume (Roberts et al, 2003).
Other Signaling Pathways
Sympathetic signaling pathways are important in the pathophysiology of LUTS, as is evident from the use of drugs interfering with the adrenergic nervous system such as α-adrenergic receptor blockers, which are highly effective for the treatment of LUTS (American Urological Association, 2003). In addition, there is increasing evidence that sympathetic pathways may be important in the pathogenesis of the hyperplastic growth process (McVary et al, 1994, 2005). α-Adrenergic blockade, in some model systems, can induce apoptosis (Anglin et al, 2002). α-Adrenergic pathways can also modulate the smooth muscle cell phenotype in the prostate (Lin et al, 2000). All the components of the renin-angiotensin system (RAS) are present in prostatic tissue and may be activated in BPH (Dinh et al, 2001, 2002; Fabiani et al, 2001). Either with or without sympathetic modulation, local RAS pathways may contribute to cell proliferation and smooth muscle contraction.
The early growth response gene-1 (EGR1) transcription regulation pathway was found to be active in a BPH cell line (Mora et al, 2005). Also of interest is the finding that α2-macroglobulin, a large protein that binds PSA and many growth factors, is very highly expressed in human prostate and is upregulated in BPH (Lin et al, 2005). Trapping and inactivation of inhibitory molecules could promote growth pathways.
Potential Role of Inflammatory Pathways and Cytokines in Benign Prostatic Hyperplasia
An additional source of growth factors in human BPH tissue may be the inflammatory cell infiltrates seen in many men with BPH. In the 1990s, descriptive studies suggested a link between inflammation and BPH-related growth. Theyer and associates (1992) reported extensive infiltration of human BPH tissues by activated T cells. Peripheral blood and tumor infiltrating T cells are known to express VEGF, a potent epithelial mitogen (Blotnik et al, 1994; Freeman et al, 1995). T cells are known to produce and secrete a variety of other growth factors, including HB-EGF and bFGF/FGF-2. Thus T cells present in the local prostate environment were thought to be capable of secreting potent epithelial and stromal mitogens that promote stromal and glandular hyperplasia.
In the past 5 years, specific inflammatory mediator pathways have been studied in detail to elucidate the potential role of these pathways in BPH pathogenesis. A large number of cytokines and their receptors are seen in BPH tissue (Konig et al, 2004). Specifically, significant levels of IL-2, IL-4, IL-7, IL-17, interferon-γ (IFN-γ), and their relevant receptors are found in BPH tissue (Kramer et al, 2002; Steiner et al, 2003a, 2003b). IL-2, IL-7, and IFN-γ stimulate the proliferation of prostatic stromal cells in vitro. Prostatic epithelial cell senescence results in increased expression of IL-8, which can promote proliferation of nonsenescent epithelial and stromal cells (Castro et al, 2004). Macrophage inhibitory cytokine-1 is expressed in normal prostate tissue but significantly downregulated in BPH (Kakehi et al, 2004; Taoka et al, 2004). Chronic inflammation in BPH is also associated with focal upregulation of cyclooxygenase 2 (COX-2) in the glandular epithelium (Wang et al, 2004). To date, however, no firm cause-and-effect relationships have been established between prostatic inflammation and related cytokine pathways and stromal-epithelial hyperplasia.
An excellent recent review of BPH as a potentially autoimmune disease was published by Kramer and colleagues (2007), and Figure 91–4 illustrates the immunologic key features of chronic inflammation in BPH and the present interpretation of these changes in the development and progression of BPH.
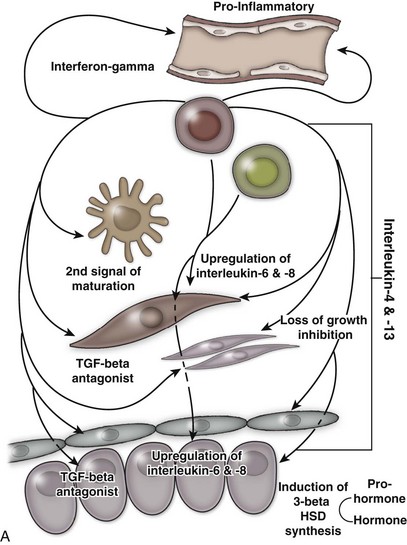
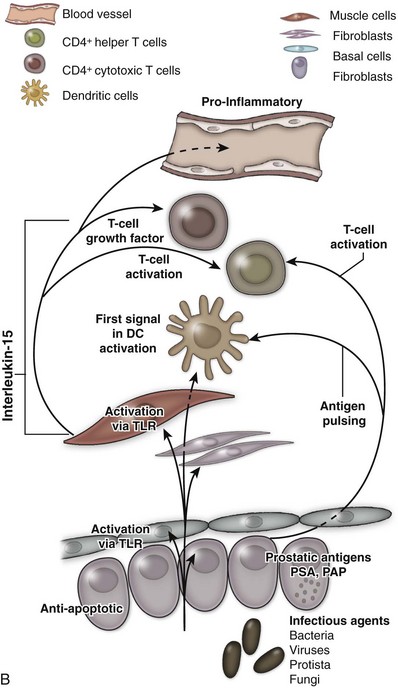
(From Kramer G, Mitteregger D, et al. Is benign prostatic hyperplasia [BPH] an immune inflammatory disease? Eur Urol 2007;51[5]:1202–16.)
Genetic and Familial Factors
There is substantial evidence that BPH has an inheritable genetic component. Sanda and colleagues conducted a retrospective case-control analysis of surgically treated BPH patients and control subjects at Johns Hopkins Hospital (Partin et al, 1994; Sanda et al, 1994). The BPH patients were men whose resected prostate weights were in the highest quartile (>37 g) and whose age at prostatectomy was in the lowest quartile. The hazard-function ratio for surgically treated BPH among first-degree male relatives of the BPH cases compared with the first-degree male relatives of the controls was 4.2 (95% confidence interval [CI], 1.7 to 10.2), demonstrating a very strong relationship (Table 91–3). The results did not appear to be due to differences in health-seeking behavior between the two groups. A segregation analysis showed that the results were most consistent with an autosomal dominant inheritance pattern. Utilizing this model, approximately 50% of men undergoing prostatectomy for BPH when younger than 60 years of age could be attributable to an inheritable form of disease. In contrast, only about 9% of men undergoing prostatectomy for BPH when older than 60 years of age would be predicted to have a familial risk. In addition, monozygotic twins demonstrate a higher concordance rate of BPH than dizygotic twins (Partin et al, 1994).
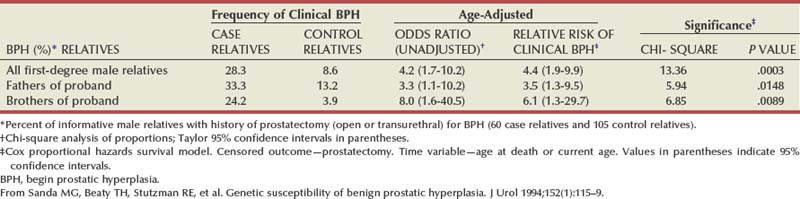
In a community-based cohort study of more than 2000 men, Roberts and colleagues (1995) found an elevated risk of moderate to severe urologic symptoms in men with a family history of an enlarged prostate and a family history of BPH compared with those with no history. Analysis of the subjects who participated in the U.S. finasteride clinical trial identified 69 men who had three or more family members with BPH, including the proband (Sanda et al, 1997). Regression analysis demonstrated that familial BPH was characterized by large prostate size, with a mean prostate volume of 82.7 mL in men with hereditary BPH compared with 55.5 mL in men with sporadic BPH. Serum androgen levels and the response to 5α-reductase inhibition were similar in familial and sporadic BPH. A more recent familial aggregation study in the finasteride database confirmed that a strong family history of early-onset and large prostate volume is more likely to be associated with inheritance of risk than symptom severity or other factors (Pearson et al, 2003).
These studies clearly demonstrate the presence of a familial form of BPH and suggest the presence of a gene contributing to the pathogenesis of the disease. The studies of Meikle and coworkers (1997, 1999) also support a genetic basis for BPH. Preliminary studies demonstrate evidence of DNA mutations (White et al, 1990), DNA hypomethylation (Bedford and van Helden, 1987), and abnormalities of nuclear matrix protein expression (Partin et al, 1993), miscellaneous genetic polymorphisms (Werely et al, 1996; Konishi et al, 1997; Habuchi et al, 2000), and abnormal expression of the Wilms tumor gene (WT1) (Dong et al, 1997) in human BPH. However, the specific gene or genes involved in familial BPH or that contribute to the risk of significant prostatic enlargement in sporadic disease remain to be elucidated.
Other Etiologic Factors
Androgens and soluble growth factors are clearly not the only important factors for the development of BPH. All mammalian prostates studied have testosterone, DHT, and AR as well as most of the known growth factor signaling pathways; however, only dog and man develop BPH. Interestingly, another glandular organ that remains androgen responsive throughout life, the seminal vesicle, does not develop hyperplasia. Obviously, other mechanisms or cofactors must be present in these two unique species making them susceptible to the disease. Nonandrogenic substances from the testis, perhaps transmitted through the vas deferens or deferential blood vessels, for example, may play some role (Dalton et al, 1990). Rats with intact testes treated with exogenous androgen demonstrate a greater degree of prostatic growth than castrated rats treated with androgen. Sutkowski and coworkers (1993) have demonstrated that human spermatocele fluid is mitogenic to both human prostatic epithelial and stromal cells in culture. Similar results have been seen in castrated versus testes-intact dogs treated with exogenous androgen and exogenous testosterone and estradiol combination (Juniewicz et al, 1994). In addition to increases in prostate weight, the incidence of histologic BPH was significantly higher in the dogs with intact testes. Grayhack and colleagues (1998) have identified a putative substance that may be a candidate for such a factor.
Prolactin has long been speculated to play a role in BPH because of the known effects of this hormone on prostate cells in vitro. Transgenic mice overexpressing the prolactin gene develop significant enlargement of the prostate (Wennbo et al, 1997). However, despite the documented presence of prolactin receptors in the human prostate and low circulating levels of the hormone, the role of prolactin in human prostate disease is unclear.
Molecular profiling, fingerprinting, microarrays, and high-throughput screening tools have uncovered new genes, as well as known genes not previously associated with BPH. Preliminary findings from the Getzenberg laboratory (Prakash et al, 2002; Sakamoto et al, 2004a, 2004b; Shah et al, 2004; Minnery and Getzenberg, 2005) and other groups (Fromont et al, 2004; Dhanasekaran et al, 2005) suggest that new markers for BPH and new therapeutic targets will be found in the next few years.
Pathophysiology
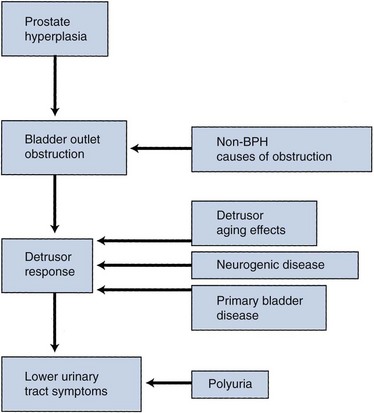
The pathophysiology of BPH is complex (Fig. 91–5). Prostatic hyperplasia increases urethral resistance, resulting in compensatory changes in bladder function. However, the elevated detrusor pressure required to maintain urinary flow in the presence of increased outflow resistance occurs at the expense of normal bladder storage function. Obstruction-induced changes in detrusor function, compounded by age-related changes in both bladder and nervous system function, lead to urinary frequency, urgency, and nocturia, the most bothersome BPH-related complaints. Thus, an understanding of BPH pathophysiology requires detailed insight into obstruction-induced bladder dysfunction.
Pathology
Anatomic Features
McNeal (1978) demonstrated that BPH first develops in the periurethral transition zone of the prostate (Fig. 91–6). The transition zone consists of two separate glands immediately external to the preprostatic sphincter. The main ducts of the transition zone arise on the lateral aspects of the urethral wall at the point of urethral angulation near the verumontanum. Proximal to the origin of the transition zone ducts are the glands of the periurethral zone that are confined within the preprostatic sphincter and course parallel to the axis of the urethra. All BPH nodules develop either in the transition zone or in the periurethral region (McNeal, 1978, 1990). Although early transition zone nodules appear to occur either within or immediately adjacent to the preprostatic sphincter, as the disease progresses and the number of small nodules increases they can be found in almost any portion of the transition or periurethral zone. However, the transition zone also enlarges with age, unrelated to the development of nodules.
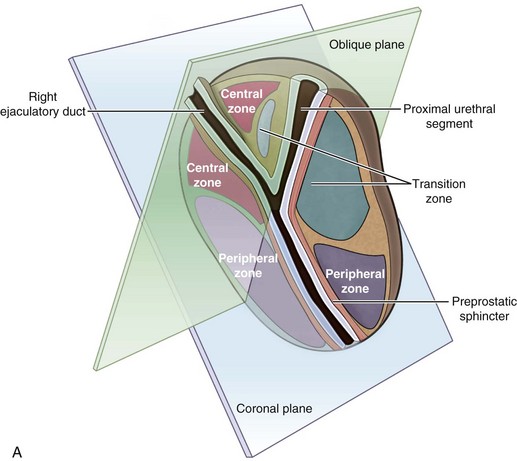
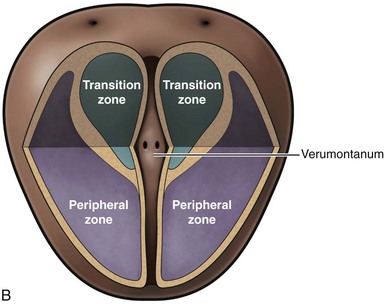
Figure 91–6
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


