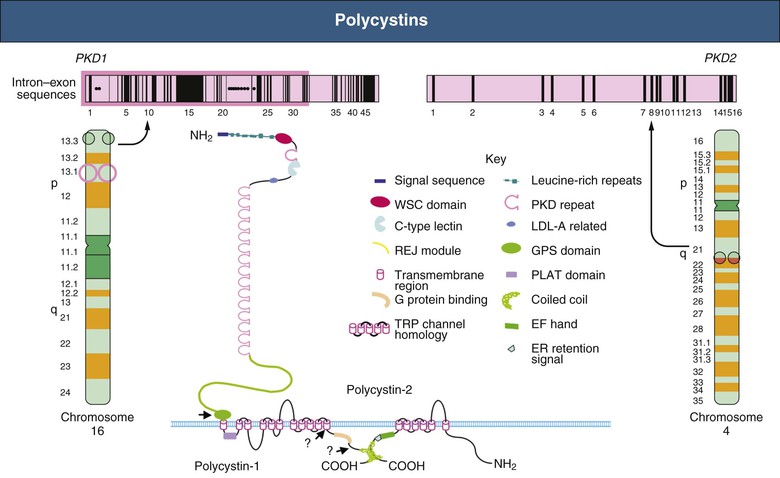
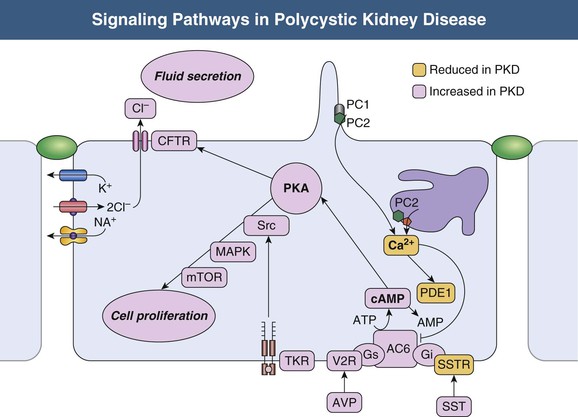
Vicente E. Torres, Peter C. Harris Autosomal dominant polycystic kidney disease is a multisystem disorder characterized by multiple, bilateral renal cysts associated with cysts in other organs, such as liver, pancreas, and arachnoid membranes.1 It is a genetic disorder caused by mutations in one of two different genes and is expressed in an autosomal dominant pattern, with variable expression. Although benign renal cysts are common, multiple bilateral cysts are not. Therefore, an underlying inherited disease should be considered in patients with normal renal function and multiple bilateral renal cysts. The autosomal dominant polycystic kidney disease (ADPKD) proteins now known as polycystin-1 and polycystin-2 play a critical role in the normal function of the primary cilium that is essential to maintaining the differentiated phenotype of tubular epithelium.2,3 Autosomal dominant polycystic kidney disease (ADPKD) is genetically heterogeneous with two genes identified (Fig. 46-1), PKD1 (chromosome 16p13.3) and PKD2 (4q21). Autosomal dominant polycystic liver disease (ADPLD) also exists as an independent entity and is genetically heterogeneous; the first two genes identified (PRKCSH in chromosome 19 and SEC63 in chromosome 6) account for about one third of isolated ADPLD cases. Evidence from animal models of ADPKD and analysis of cystic epithelia have shown that renal cysts may develop from loss of functional polycystin with somatic inactivation of the normal allele. However, cysts can develop even if the protein is not completely lost, as demonstrated by animal models expressing incompletely penetrant alleles.4 Furthermore, transgenic rodents overexpressing Pkd1 or Pkd2 develop renal cystic disease, which suggests that multiple genetic mechanisms causing an imbalance in expression of polycystins can lead to the development of cysts.2,3 Polycystin-1 (PC1) and polycystin-2 (PC2) belong to a subfamily of transient receptor potential (TRP) channels. PC1 (TRPP1; ~460 kd) has the structure of a receptor or adhesion molecule and contains a large extracellular N region, 11 transmembrane regions, and a short intracellular C region (Fig. 46-1). PC1 interacts with PC2 through a coiled-coil domain in the C-terminal portion and with multiple other proteins at different extracellular and intracellular sites. PC1 is found in the primary cilia, cytoplasmic vesicles, plasma membrane at focal adhesions, desmosomes, adherens junctions, and possibly endoplasmic reticulum and nuclei. PC1 may regulate the mechanical strength of adhesion between cells by controlling the formation of stabilized, actin-associated adherens junctions. PC2 (TRPP2; ~110 kd) contains a short N-terminal cytoplasmic region, six transmembrane domains, and a short C-terminal portion. PC2 is localized predominantly to the endoplasmic reticulum but also to the plasma membrane, primary cilium, centrosome, and mitotic spindles in dividing cells.2,3 PC1 and PC2 are also found at high concentrations in exosomes, which are shed into the urine and physically interact with primary cilia, possibly exerting a urocrine function of cell-cell communication.5 Experimental data indicate that the timing of ciliary loss or Pkd1 inactivation determines the rate of development of cystic disease. Inactivation in the developing kidney results in rapid progression.6 Cysts also develop rapidly in the corticomedullary region (pars recta and thick ascending limbs of Henle loop) of adult mouse kidneys subjected to ischemic injury to stimulate cell proliferation.7 The polycystins are involved in the detection of extracellular cues at primary cilia, cell-cell contacts, and cell-matrix contacts and are essential to maintain the differentiated phenotype of the tubular epithelium. Reduction in one of the polycystins below a critical threshold results in inability to maintain planar polarity, increased rates of proliferation and apoptosis, expression of a secretory phenotype, and remodeling of the extracellular matrix.2,3 PC1 and PC2 in the primary cilium are required for a calcium transient in response to ciliary bending.8 PC2 is a TRP channel (TRPP2), and functions as a calcium release channel in the endoplasmic reticulum.9 PC1 interacts with and modulates the function of PC2. PC1 and PC2 also interact with additional calcium channel proteins. Precisely how intracellular calcium homeostasis is altered in ADPKD remains uncertain, but many studies show reduced resting intracellular calcium, endoplasmic reticulum calcium stores, and store-operated calcium entry in primary cell cultures or microdissected samples from human and rodent polycystic tissues2–3 (Fig. 46-2). A common finding in animal models of PKD is increased levels of cyclic adenosine monophosphate (cAMP), not only in the kidney but also in the liver and vascular smooth muscle.10 Tissue levels of cAMP are determined by the activities of membrane-bound and soluble adenylyl cyclases (ACs) and cAMP phosphodiesterases (PDEs), themselves subject to complex regulatory mechanisms. Reduced intracellular calcium in PKD may activate calcium inhibitable AC6, directly inhibit calcium/calmodulin-dependent PDE1, and indirectly inhibit cGMP inhibitable PDE3, therefore accounting for the accumulation of cAMP and activation of protein kinase A (PKA), which in turn contributes to the development and progression of PKD by stimulating CFTR-driven chloride and fluid secretion and cell proliferation (Fig. 46-2). Chloride enters across basolateral Na-K-2Cl cotransporters, driven by the sodium gradient generated by basolateral Na+,K+-ATPase, and exits across apical PKA–stimulated cystic fibrosis transmembrane conductance regulator (CFTR). Basolateral recycling of potassium occurs through the KCa3.1 channel. Cyclic AMP exerts opposite effects on cell proliferation in different cell types. Cyclic AMP and PKA signaling enhance several pro-proliferative pathways (extracellular signal–regulated kinase, ERK) in cells derived from polycystic kidneys, while inhibiting proliferation in cells derived from normal human kidney cortex.11,12 Treatment of normal human kidney or murine collecting duct cells with calcium channel blockers replicates the proliferative response of the ADPKD cells to cAMP, thus linking this response to the reduction in intracellular calcium that results from disrupting the polycystin pathway.13 Conversely, treatment of ADPKD cyst derived cells with calcium channel activators or calcium ionophores restores the normal antimitogenic response to cAMP (Fig. 46-2). Activation of mTOR signaling downstream from PKA through ERK-mediated phosphorylation of tuberin has been linked to transcriptional activation of aerobic glycolysis, increased levels of adenosine triphosphate (ATP), and, together with ERK-dependent inhibition of liver kinase B1 (LKB1) and inhibition of AMP kinase (AMPK).14–16 Liver cysts arise by excessive proliferation and dilation of biliary ductules and peribiliary glands. Estrogen receptors, insulin-like growth factor 1 (IGF-1), IGF-1 receptors, and growth hormone receptor are expressed in the epithelium lining the hepatic cysts, and estrogens and IGF-1 stimulate hepatic cyst–derived cell proliferation.17 Cyst growth is also promoted by growth factors and cytokines secreted into the cyst fluid. Hypertension is a major clinical manifestation and predictor of outcome in ADPKD (see Clinical Manifestations). Several factors contribute to the development of hypertension in ADPKD. Activation of the intrarenal renin-angiotensin system (RAS) likely plays an important role, but controversy surrounds whether the circulating RAS is inappropriately activated.18 The expression of PC1 and PC2 in vascular smooth muscle and endothelium, along with enhanced vascular smooth muscle contractility and impaired endothelium-dependent vasorelaxation in ADPKD, suggest that disruption of polycystin function directly contributes to hypertension. Other factors include increased sympathetic nerve activity and plasma endothelin-1 levels and insulin resistance.19 Endothelial vasodilation and constitutive nitric oxide synthase (NOS) activity are reduced in subcutaneous resistance vessels from patients with ADPKD and normal glomerular filtration rate (GFR). Flow-induced vasodilation of the brachial artery is inconsistently impaired, whereas pulse wave reflection is amplified, suggesting a predominant involvement of small resistance vessels. Reduced coronary flow velocity reserve and increased carotid intima-media thickness in normotensive patients with normal GFR suggest that atherosclerosis starts early in the course of ADPKD.20 Reduced nitric oxide (NO) endothelium-dependent vasorelaxation in ADPKD may be caused by increased plasma levels of asymmetric dimethylarginine, a mechanism common to all hypertension associated with kidney disease (see Chapter 82). Autosomal dominant PKD occurs worldwide and in all races, with a prevalence of genetically affected individuals at birth estimated at 1 : 400 to 1 : 1000.1 In most patients, however, the diagnosis is made decades later, and some patients are never diagnosed. Therefore, at any point in time, only a fraction of genetically affected individuals are aware of having the disease. The percentage of end-stage renal disease (ESRD) caused by ADPKD is less among African Americans than among Caucasians because of a higher incidence of other causes of ESRD. Yearly incidence rates for ESRD caused by ADPKD in men and women, respectively, are 8.7 and 6.9 per 1 million (1998 to 2001, United States), 7.8 and 6.0 per million (1998 and 1999, Europe), and 5.6 and 4.0 per million (1999 and 2000, Japan). Age-adjusted gender ratios greater than unity (1.2 to 1.3) suggest more progressive disease in men than in women. In recent studies, the age of patients with ESRD has increased in both genders; the age-adjusted male-female ratio for onset of ESRD has trended toward unity; and all-cause mortality has decreased, possibly because of improved detection and control of hypertension.21,22 Genic, allelic, and gene-modifier effects contribute to the high phenotypic variability of ADPKD. PKD1-associated disease is more severe than PKD2-associated disease (age at ESRD, 54 years vs. 74 years for PKD1 and PKD2, respectively). The greater severity of PKD1 is caused by development of more cysts at an early age, not faster cyst growth.23 Both PKD1 and PKD2 can be associated with severe polycystic liver disease and vascular abnormalities. Because of the lesser severity of the renal involvement, the prevalence of PKD2-associated disease has likely been underestimated in clinical studies. Mutations in PKD1 and PKD2 are highly variable and usually “private” (unique to a kindred). The ADPKD Mutation Database (http://pkdb.mayo.edu) lists 868 truncating PKD1 mutations identified in 1243 families with a total of 2322 variants, including missense mutations and silent polymorphisms. Also, 162 PKD2 truncating mutations are listed in 278 families, with a total of 374 different variants. Allelic factors (mutation type or location) have an effect on the severity of ADPKD. Patients with mutations in the 5′ region of PKD1 were found to have more severe disease than patients with 3′ mutations. More recent studies in larger cohorts have shown that the type of PKD1 mutation, but not its position, correlates strongly with renal survival. The median age at onset of ESRD was 55 years for carriers of a truncating mutation and 67 years for carriers of a nontruncating mutation.24 Hypomorphic or incompletely penetrant PKD1 or PKD2 alleles have been described.25 These alleles alone may result in mild cystic disease; two such alleles cause typical to severe disease and in combination with an inactivating allele, may be associated with early-onset disease that mimics ARPKD.26 The large intrafamilial variability of ADPKD highlights a role for genetic background in disease presentation. Age at clinical manifestations in ADPKD is less variable within than between families, which suggests a common familial modifying background for early and severe disease expression (e.g., mutations or variants in genes encoding other cystoproteins). The contiguous deletion of the adjacent PKD1 and TSC2 is characterized by childhood PKD with additional clinical signs of tuberous sclerosis complex. Other modifying loci are likely to account for more common and subtle intrafamilial variability. Only individuals who have been properly informed about the advantages and disadvantages of screening should be offered presymptomatic screening. If ADPKD is diagnosed, the patient should receive appropriate genetic counseling, and risk factors such as hypertension can be identified and treated early. If ADPKD is absent, the patient can be reassured. Disadvantages of presymptomatic screening relate to insurability and employability. Presymptomatic screening of children is not recommended, but this advice will likely change when more effective therapy for the disease becomes available. Renal ultrasound is used for presymptomatic testing because of cost and safety. Revised criteria have been proposed to improve the diagnostic performance of ultrasound in ADPKD (Table 46-1). At least three (unilateral or bilateral) renal cysts and two cysts in each kidney are sufficient for diagnosis of at-risk individuals age 15 to 39 and 40 to 59 years, respectively.27 For at-risk individuals age 60 and older, four or more cysts in each kidney are required. Table 46-1 Ultrasound criteria for diagnosis of autosomal dominant polycystic kidney disease (ADPKD). NPV, Negative predictive value; PPV, positive predictive value. (From reference 61.) Whereas the overall specificity and positive predictive value (PPV) of ultrasound are high by use of these criteria, their sensitivity and negative predictive value (NPV) when applied to PKD2 patients age 15 to 59 are low. This is a problem in the evaluation of potential kidney donors, in which exclusion of the diagnosis is important.17 Different criteria have therefore been proposed to exclude a diagnosis of ADPKD in an individual at risk from a family with an unknown genotype. An ultrasound finding of normal kidneys or one renal cyst in an individual age 40 or older has an NPV of 100%. The absence of any renal cyst provides near certainty that ADPKD is absent in at-risk individuals age 30 to 39, with a false-negative rate of 0.7% and NPV of 98.7%. A normal or indeterminate ultrasound scan does not exclude ADPKD with certainty in an at-risk individual younger than 30; normal magnetic resonance imaging (MRI) or contrast-enhanced computed tomography (CT) provides further assurance, but data are insufficient to quantify its predictive accuracy. Genetic testing can be performed when a precise diagnosis is needed and the results of imaging are indeterminate. However, there are limitations to genetic testing, by either linkage or mutation analysis. Linkage analysis requires accurate diagnosis and the availability and willingness of sufficient affected family members to be tested; it is feasible in less than 50% of families. De novo mutations can also complicate interpretation of results. Molecular testing by direct DNA sequencing is now informative in about 90% of patients.28 However, because most mutations are unique and up to one third of PKD1 changes are missense, the pathogenicity of some changes is difficult to prove. In preimplantation genetic diagnosis, genetic analysis is performed on single blastomeres from preimplantation embryo biopsy specimens obtained after in vitro fertilization (IVF), and only embryos unaffected by the disease are selected for transfer. Preimplantation genetic diagnosis for ADPKD is complicated by the genetic heterogeneity of the disease and by the large size and complex structure of the PKD1 gene. Demand is limited in this late-onset disorder, and it has been performed in very few cases. Renal cystic disease can be a manifestation of many other systemic diseases. Conditions to consider when renal cystic disease is detected but presentation is not typical of ADPKD include autosomal recessive PKD, tuberous sclerosis complex, von Hippel–Lindau disease, renal cysts and diabetes (RCAD) from HNF1β mutations, and orofaciodigital syndrome type I, as well as medullary sponge kidney and simple renal cysts. These are discussed further, including differential diagnosis, in Chapter 47. If the patient has ESRD, acquired cystic disease should also be considered (see Chapter 89). Autosomal dominant PKD is a multisystem disorder. Multiple renal and extrarenal manifestations of ADPKD have been described that cause significant complications. A number of clinical features that result from renal damage can be identified (Box 46-1). Reduction in urinary concentrating capacity and glomerular hyperfiltration are early functional abnormalities that can be observed in some children and adolescents with ADPKD. Renal size increases with age, and renal enlargement eventually occurs in 100% of patients with ADPKD. The severity of the structural abnormality correlates with the manifestations of ADPKD, such as pain, hematuria, hypertension, and renal impairment.29 Massive renal enlargement can lead to compression of local structures, resulting in such complications as inferior vena cava (IVC) compression and digestive symptoms. Most manifestations are directly related to the development and enlargement of renal cysts. The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP), a prospective study of 241 patients by annual MRI, has shown that total kidney volume and cyst volumes increased exponentially.30 Rates of growth were relatively constant, averaging 5.3% per year, but highly variable from patient to patient. Baseline total kidney volume predicted the subsequent rate of increase in renal volume and decline in GFR.31 Episodes of acute renal pain are seen frequently; causes include cyst hemorrhage, infection, stone, and rarely tumor, and these must be investigated thoroughly. A few ADPKD patients with renal enlargement and structural distortion develop chronic flank pain without specifically identifiable etiology. Visible hematuria may be the initial presenting symptom and occurs in up to 40% of ADPKD patients over the course of the disease. Many have recurrent episodes. Differential diagnosis includes cyst hemorrhage, stone, infection, and tumor. Cyst hemorrhage is a frequent complication and produces gross hematuria when the cyst communicates with the collecting system. Frequently, the cyst does not communicate with the collecting system, and flank pain without hematuria occurs. It can present with fever, raising the possibility of cyst infection. On occasion, a hemorrhagic cyst will rupture, resulting in a retroperitoneal bleed that can be significant, potentially requiring transfusion. In most patients, cyst hemorrhage is self-limited, resolving within 2 to 7 days. If symptoms of hematuria or flank pain last longer than 1 week or if the initial episode of hematuria occurs after age 50 years, investigation to exclude neoplasm should be undertaken.
Autosomal Dominant Polycystic Kidney Disease
Definition
Etiology and Pathogenesis
Genetic Mechanisms
Polycystic Kidney Disease Proteins
Mechanisms of Cyst Formation
Liver Cyst Development
Hypertension
Epidemiology
Phenotypic Variability
Diagnosis
Renal Ultrasound
Ultrasound Criteria for the Diagnosis of ADPKD
Age (years)
Criteria
PPV
NPV
Original Ravine’s PKD1 Diagnostic Criteria
15-29
≥2 cysts, unilateral or bilateral
99
88
30-39
≥2 cysts in each kidney
100
88
40-59
≥2 cysts in each kidney
100
95
≥60
≥4 cysts in each kidney
100
100
Revised Unified Diagnostic Criteria
15-29
≥3 cysts, unilateral or bilateral
100
86
30-39
≥3 cysts, unilateral or bilateral
100
86
40-59
≥2 cysts in each kidney
100
95
≥60
≥4 cysts in each kidney
100
100
Revised Criteria for Exclusion of Diagnosis
15-29
≥1 cyst
97
91
30-39
≥1 cyst
94
98
40-59
≥2 cysts
97
100
≥60
≥3 cysts in each kidney
100
100

Genetic Testing
Differential Diagnosis
Clinical Manifestations
Renal Manifestations
Renal Size
Pain
Hematuria and Cyst Hemorrhage
Urinary Tract Infection and Cyst Infection
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Autosomal Dominant Polycystic Kidney Disease
Chapter 46