Fig. 26.1
Mechanisms of allograft injury by donor-specific antibody
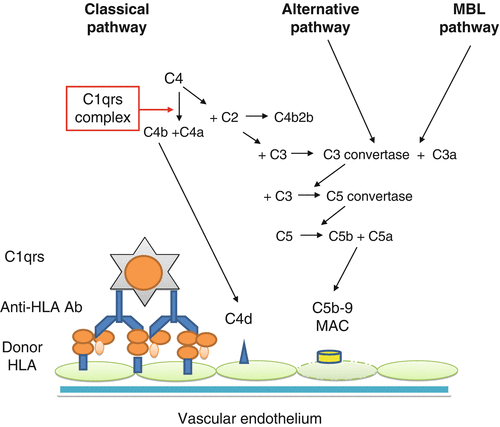
Activation of complement system plays a major role in antibody-mediated graft injury. After the binding of DSA to and cross-linking of target epitopes on the surface of endothelial cells, this system becomes activated through classical pathway (Fig. 26.2). With final activation of C5, membrane attack complex (MAC) will form by assembly of C5b-C9, which can cause cell apoptosis and death. The by-product of activation of complement system is the formation of anaphylatoxins (C3a, C5a) that cause chemotaxis and activation of inflammatory cells [21]. C4d is the breakdown product of C4b, with unclear biological function that can bind covalently to the surface of endothelial cells and basement membrane. In normal kidneys, C4d can be found in the glomerular vascular pole and mesangium, due to the high complement turnover. In cases of immune-mediated glomerulopathies, C4d deposition can extend into the glomerular capillaries, but only rarely into the PTC [22, 23]. Its detection in PTC generally indicates antibody-mediated activation of complement system; therefore, it is considered the “fingerprint” telltale sign of the antibody action. However, not all cases with AMR demonstrate PTC C4d deposition. Halloran and colleagues have provided evidence for C4d-negative AMR [24–26]. Complement-independent antibody-mediated endothelial injury has been clearly shown in animal models [27]. Recent experimental evidence suggests the important role of natural killer (NK) cells, independent of complement, in antibody-mediated chronic allograft vasculopathy in murine heart allografts [28]. Histological and gene expression profiling of renal allograft biopsies also support the role of NK cells in pathogenesis of AMR [26].
The endothelial response to DSA depends on different factors including density of the antigen on the cells, affinity of the antibody for the target, its complement-activating ability, concentration of antibody, synergy between antibodies against different epitopes [30], and presence of complement regulatory proteins. IgG3 and IgG1 strongly activate complement, while IgG2 is a weak and IgG4 is a poor complement activator [31, 32].
At higher concentrations, DSA binding can lead to lytic lesions and apoptosis of the cells with denudation of the underlying matrix and subsequent platelet activation and thrombosis in the microvasculature [33–36]. The lytic changes are generally found in the early posttransplant period [33, 37–39] and can vary in severity from mild to severe [40]. However, at lower concentrations, antibody ligation of HLA molecules on the endothelial cells stimulates cell proliferation, with upregulation of fibroblast growth factor receptor and activation of mTOR pathway playing important roles [41, 42]. The resultant endothelial cell activation is accompanied by increased expression of adhesion molecules VCAM-1 and ICAM-1 and release of von Willebrand factor and P-selectin, leading to adherence and activation of inflammatory cells and platelets [42]. Sublytic levels of DSA can cause rearrangement of the actin cytoskeleton leading to change in endothelial cell morphology from polygonal and flat to contracted and rounded. This transformation opens gaps between the cells to expose the underlying matrix, triggering activation of the coagulation cascade and platelet aggregation, resulting in formation of microthrombi [37, 43]. The activated platelets in turn recruit inflammatory cells by expressing thromboxanes and RANTES [44, 45] and cause the expression of P-selectin and integrins that aid leukocyte trafficking [46, 47].
Presence of anti-graft antibody without functional injury has been postulated (accommodation). This phenomenon is more common with ABO antibodies [48–52]. Upregulation of complement regulatory proteins, decay accelerating factor, complement receptor-related protein, and CD59 is one of the hypothesized mechanisms of accommodation. Increased expression of pro-survival genes such as Bcl-2, Bcl-xL, and cytoprotective heme oxygenase and nitric oxide could also be involved [25, 42, 48, 53–58].
Study of gene expression patterns in the biopsies has shown that grafts with AMR have increased expression of endothelial cell transcripts [26, 59]. The presence of this expression pattern in biopsy along with DSA is significantly associated with higher risk of graft failure.
DSA: Epidemiology and Impact on Outcome
Sensitization to HLA antigens and other non-HLA antigens can happen with pregnancy, previous organ transplantation, blood transfusion, or through cross-reactivity with infectious agents or allergens. The degree of sensitization is assessed pre-transplant by using PRA assay. Rising PRA value indicates that the recipient has antibodies against larger number of HLA antigens, either from prior direct exposure or through epitope sharing. Currently, 34.6 % of patients on the US transplant wait list are sensitized to HLA antigens, 15.5 % with peak PRA >80 % (Table 5.1a, US Registry of Transplant Recipients 2010) [60]. Crossmatch testing prevents transplanting an organ in a recipient with DSA at a high enough titer and affinity to make the assay positive that could lead to hyperacute rejection. However, low level DSA could be unrecognized or even accepted for transplantation. In addition, the recipients may develop DSA de novo or by anamnestic response from prior exposure.
There are a number of studies that have examined the impact of pre-transplant and de novo DSA on long-term outcome. The available data suggest that DSA is generally predictive of worse results, including graft function, proteinuria, and graft failure.
In a study on 402 patients with negative CDC crossmatch at transplant, 8-year graft survival was significantly worse (61 %) among patients with preexisting HLA-DSA compared with both sensitized patients without HLA-DSA (93 %) and nonsensitized patients (84 %) [61]. Close to 40 % of those with pre-transplant DSA experienced AMR. The peak HLA-DSA Luminex mean fluorescence intensity (MFI) predicted AMR better than current HLA-DSA MFI (P = 0.028). Amico et al. showed a significantly higher risk of AMR or ACR in patients with pre-transplant DSA [62]. However, only those experiencing AMR had worse graft survival. In another study, Couzi et al. reported that the presence of HLA-DSA detected only by the single antigen bead (SAB) assay in the context of a negative flow cytometry (FC) crossmatch was not associated with increased risk of acute rejection [63]. In a cohort of 54 patients with preformed DSA who received adjunctive IVIG with or without rituximab and plasma exchange, 80 % experienced subclinical AMR at 3 months; this group had a higher rate of interstitial fibrosis and tubular atrophy (IFTA) (100 % vs. 33.3 %, P < 0.01) and a higher rate of transplant glomerulopathy (TG) (43 % vs. 0 %, P = 0.02) compared to patients without subclinical AMR at 3 months [64]. More recently, in a systematic review and meta-analysis, Mohan et al. concluded that the presence of preformed DSA, despite a negative FC crossmatch, nearly doubles the risk of AMR (RR: 1.98, P < 0.001, 95% CI: 1.36–2.89) and increases the risk of graft failure by 76 % (P = 0.01, 95% CI: 1.13–2.74) [65].
As noted previously, DSA can develop de novo. In a prospective study of more than 1,200 patients, 5.5 % developed donor-specific HLA and 11.3 % developed non-donor-specific HLA antibodies de novo [66]. In a smaller cohort of 249 recipients, 11.2 % developed HLA antibodies, and 4.4 % had DSA [67]. HLA antibody in this cohort was associated with higher serum creatinine. In a report from a UK center, among patients transplanted between 1985 and 2006 in those who developed DSA, 3-year graft survival after antibody detection was worse (69.5 % vs. 91.1 %, P = 0.035) [68]. Graft failure was more likely in patients with DSA with excess of 0.2 g/L proteinuria at antibody detection (42 % vs. 0 %, P = 0.008). In a cohort of 244 consecutively transplanted kidney and kidney/pancreas recipients without pre-transplant DSA transplanted between 2007 and 2009 and screened for de novo DSA at 1, 6, 12, and 24 months and when clinically indicated, DSA was detected in 27 % of all patients, all during the first year posttransplant [69]. DSA-positive patients were significantly more likely to have acute rejection (both AMR/ACR) compared with no DSA (29 % vs. 9.5 %, P < 0.001) and lower estimated 2-year graft survival (83 % vs. 98 %, P < 0.001). Ntokou et al. demonstrated de novo DSA in 15.4 % and third party HLA antibody in 32.8 % of the patients in Greece [70]. Presence of HLA antibody was an independent predictor of graft failure: for class I (HR: 31.8), class II (HR: 20.9), and non-DSA HLA antibody (HR: 5.9).
Detection of DSA with graft dysfunction or for-cause biopsies also has been shown to be associated with worse graft outcome. Our group analyzed 297 patients who underwent for-cause biopsies; 31.3 % were DSA-positive, 58 (19.5 %) with class I, 68 (22.9 %) with class II DSA, and 33 (11.1 %) with both class I and class II DSA [71]. Those with class I only DSA did not have higher risk of graft failure from the time of sampling compared with DSA-negative patients, while those with class II DSA alone or in combination with class I DSA had worse graft survival (adjusted HR: 2.5, P = 0.001, 95% CI: 1.4–4.3 for class II only and HR: 2.7, P < 0.001, 95% CI: 1.6–4.7 for class I/II DSA). Examining the time of testing posttransplant, during the first year, any form of DSA, including class I alone, was predictive of worse graft survival. However, after the first year, only class II DSA was associated with higher risk of graft failure. Lee et al. in a case-control study also showed that those with HLA antibody in first year had higher risk of graft loss (adjusted HR: 7.8, P < 0.001, 95% CI: 3.0–20.1) [72].
Despite all these reports, the presence of DSA is not always associated with poor outcome. There are reports of patients with de novo DSA who maintain stable, good graft function with no significant proteinuria for years, without obvious adverse clinical consequences [73].
Non-HLA-DSA
Clinical and histological features of AMR occur in association with circulating HLA-DSA. However, there are patients with similar presentation, but HLA-DSA cannot be identified, even using SAB assay [74]. We have previously reported that in patients with diffuse PTC C4d staining, 31.1 % did not have detectable HLA-DSA [71].
In a subset of these patients, low concentration of circulating antibody and absorption of the DSA in the allograft could explain the negative test results. It has been shown that after nephrectomy in patients with failed organ and negative DSA in the blood, the antibody can be eluted from the tissue and detected in the blood after removal of the allograft [75]. Marrari and Duquesnoy used micro bead-based Luminex antibody-binding assay in 65 cases that underwent transplant nephrectomy following rejection, before and after surgical removal of the allograft [76]. The incidence of DSA reactivity pre- and post-nephrectomy for HLA-A,B and HLA-DRB1 mismatch categories were 64 % vs. 87 % (P = 0.003) and 57 % vs. 86 % (P < 0.0001), respectively.
HLA typing for non-ABC/DR loci has not been routine. Therefore, HLA-DSA targeting DQ, DP, or DRB3-4 antigens cannot be discovered, unless the typing is performed. In a study of 138 patients with functioning grafts, DP antibody was detected in 5.1 % of the patients [77]. In contrast, among 185 patients with rejection, this frequency was 19.5 %. Jolly et al. reported two cases with pre-transplant HLA-DP DSA who after transplantation experienced recurrent acute AMR and graft failure [78]. Zhu et al. conducted a case-control study comparing 29 patients with graft failure and 25 controls with functioning grafts [79]. In the former group, out of 17 patients who received DQ-mismatched organs, 13 (76 %) had DSA against HLA-DQ antigen, compared with 1 of 11 (9 %) in the control group (P < 0.001).
Non-HLA antigens could also be the target of humoral alloimmune response. Major histocompatibility complex class I chain-related gene (MICA) DSA and anti-angiotensin receptor I (AT1) antibody are two major examples of such antibodies [80–82]. In a study of 39 rejected compared with 26 functioning grafts, IgG HLA plus MICA antibodies were found in 77 % of the former, as compared with 42 % of the latter group (P < 0.01); IgG/IgM HLA antibodies were detected in 72 % and 46 %, respectively [83]. Among 185 consecutive renal transplant patients, Panigrahi et al. detected MICA antibodies in 16 %, which was associated with worse graft survival compared to those with negative testing (60 % vs. 86 %, P = 0.001) [84]. Among the HLA antibody-negative patients, 82 % of the former and 21.4 % of the latter groups had MICA antibody. Amico et al. described ten patients with early AMR due to non-HLA-DSA; only in three, the antibody could be identified as MICA-DSA [85]. Dragun et al. for the first time reported 16 patients with refractory vascular rejection and malignant hypertension without anti-HLA antibody who had activating IgG antibodies targeting angiotensin receptor 1 (AT1) [80]. Interestingly, AT1 antagonist losartan blocked the agonistic effects of the antibodies. Other allo- or autoantibodies described as targets of antibody response in AMR causing graft injury include vimentin, agrin, collagen V, K-α1-tubulin, and nucleolin, among others [86–90]. Despite these identified targets, in some cases with clinical and pathological manifestation of AMR, the target antigen cannot be identified [34].
Regardless of the target, development of DSA is usually associated with graft injury. When this injury is acute and associated with acute rise in serum creatinine, the resultant syndrome is called acute AMR, which is associated with certain histological features. However, the antibody-mediated effect can cause slow histological changes presenting with slowly worsening graft function and proteinuria. This clinical syndrome, chronic AMR, has certain histological characteristics that separate it from acute AMR. In the next sections, we will discuss the clinical presentation, histological features, and treatment of these two syndromes.
Acute Antibody-Mediated Rejection
Introduction
Acute AMR is a clinical syndrome defined by acute graft dysfunction resulting from the effect of DSA on the allograft. This dysfunction could be in the form of acute rise in serum creatinine or undeclining serum creatinine in early posttransplant period. The extreme form of this syndrome is hyperacute rejection, in which the transplanted kidney becomes ischemic and necrotic immediately or shortly after implantation. Histologically, the allograft shows platelet aggregation along the glomerular and peritubular capillary microcirculation, leading to occlusive microthrombi and renal cortical necrosis [35, 91]. There is also pronounced neutrophilic margination and inflammation resulting in glomerulitis and peritubular capillaritis [1, 2, 92, 93]. This catastrophic event occurs as a result of high levels of undetected DSA or blood group (ABO) isoagglutinins at the time of transplantation [71, 94]. With the introduction and advances in crossmatch assays, this phenomenon has been effectively eliminated.
In 1990, Halloran et al. described a form of AMR that was different from hyperacute and typical acute rejection [40]. They described seven pre-sensitized patients with class I HLA-DSA in association with atypical but distinctive patterns of rejection. The clinical presentation was rapid onset of oliguria and deterioration of graft function, with histological picture of acute tubular necrosis. Renal histology showed granular complement deposition, polymorphonuclear infiltration, and endothelial injury in the microvasculature. In three patients, the glomerular capillaries showed fibrin thrombi and widening of the subendothelial space, similar to hemolytic uremic syndrome. They hypothesized that anti-class I antibody appearing or persisting in the early posttransplant period can cause a unique form of acute rejection characterized by prominent microvascular injury, ATN, PMN infiltration, and complement staining. During the subsequent years, this pattern of acute rejection was better characterized, and the time course of the cellular and molecular events was delineated [16]. The prominent microvascular inflammation has been consistently associated with AMR [36, 41, 56, 95–102]. Unlike hyperacute rejection, acute AMR causes a slower vascular compromise to the allograft microcirculation, hence does not present as catastrophic graft failure and loss; although uninterrupted acute AMR can progress to graft loss within a short period of time [103–105].
Clinical Presentation and Histology
Acute AMR typically presents in early posttransplant period, either as delayed or slow graft function or acute deterioration of graft function after initial improvement and stabilization. The DSA is generally anti-donor HLA antibody, but it can have a non-HLA target, as described previously. The former can be against class I or class II HLA antigen(s). In early acute AMR, class I antigens are more commonly involved. The early cases are generally the result of low DSA titers that do not cause positive crossmatch testing at the time of surgery or are undetectable at the time. Later cases are more often the result of de novo DSA.
The true incidence of acute AMR varies depending on the characteristics of the patient population, with reported rates from 5 to 23 % in unselected recipients and 12–37 % of biopsies taken for acute rejection and up to 60 % in desensitized patients with positive crossmatch or ABO-incompatible transplantation [112–118]. Histological features of AMR in the presence of DSA can be found in surveillance biopsies in the absence of graft dysfunction. These subclinical cases will increase the true rate of acute AMR after kidney transplantation.
The risk factors for acute AMR include previous transplantation, particularly those with early graft loss, multiple repeat mismatches, multiple preformed DSA, multiple sensitizing events, and high-risk combinations (husband-to-wife and child-to-mother) [119, 120].
Acute AMR as a pathological diagnosis was formally recognized in the Banff working group has revised these criteria a few times and is in the process of refining them further following the recent meeting in 2013 [106–112]. Diagnosis of acute AMR requires both morphologic evidence of graft injury and immunohistologic evidence of antibody effect. In the presence of serologic evidence, circulating DSA, this constellation can be called acute AMR (Table 26.1). According to the Banff criteria, the histological evidence can be one of the three types:
Table 26.1
Banff criteria for diagnosis of acute AMR
1. Morphologic evidence |
• Acute tubular injury (type I) |
• Neutrophils and/or monocytes/macrophages in PTC and/or glomeruli (type II) |
• Thrombi in glomerular capillaries, arterioles, and/or small arteries (type II) |
• Arterial fibrinoid necrosis (type III) |
2. Immunohistologic evidence |
• Diffuse C4d in PTC |
• Ig and/or complement in arterial fibrinoid necrosis |
3. Serologic evidence |
• Circulating DSA to donor HLA or other targets |
Type I, acute tubular injury; type II, neutrophils and/or monocytes/macrophages in PTC and/or glomeruli or thrombi in glomerular capillaries, arterioles, and/or small arteries; and type III, arterial fibrinoid necrosis. It is clear that when AMR is characterized with only acute tubular injury in the biopsy, in the absence of other supportive evidence, its distinction from other forms of tubular injury would be impossible. In other words, in patients with delayed graft function, which is generally associated with tubular injury attributed to ischemia/reperfusion injury, diagnosis of acute AMR can be missed if screening for serologic and immunohistologic markers of AMR is not carried out.
The immunohistologic evidence of AMR is documented by demonstration of C4d deposition in PTC or Ig or complement in fibrinoid necrosis. C4d can be detected either by immunofluorescence (IF) or immunohistochemistry (IHC). IF is performed on frozen tissue section using a monoclonal antibody against C4d. For IHC paraffin-embedded tissue section is stained with tagged polyclonal anti-C4d antibody [121]. For diagnosis of AMR diffuse (>50 %) PTC staining is required (Banff C4d3). Since IHC is less sensitive than IF, in more than half of the biopsies with focal pattern (10–50 % of PTC, Banff C4d2) with IHC, the pattern is diffuse using the latter [111, 121, 122]. Our group, along with others, has shown that focal staining with IHC has an association with graft survival similar to diffuse pattern [71].
The Banff classification recognizes the intensity of peritubular capillaritis (ptc) and glomerulitis (g) by scoring them from 0 to 3, based on the extent of capillaries and number of luminal inflammatory cells involved [107, 111]. ptc/g score >0 is indicative of histological evidence of AMR. Acute AMR often occurs in combination with features of ACR, including interstitial inflammation and tubulitis. Therefore, screening for DSA and C4d staining are essential for complete evaluation of an allograft biopsy.
Management
Despite the great progress that has been made in prevention and treatment of cell-mediated rejection, the treatment of AMR remains for the most part nonspecific and suboptimal. The proposed treatment strategies are mostly based on retrospective data in small cohort of patients, often with variable diagnostic criteria and with relatively short follow-up periods [123]. Diagnosis of acute AMR is based on graft dysfunction associated with histological and immunohistologic evidence of demonstrable circulating DSA. Treatment protocols for these cases are based on three basic concepts:
1.
Suppression of T-cell function to decrease/eliminate T cell-mediated B-cell activation
2.
Removal or neutralization of circulating DSA and complement components
3.
Prevention of DSA formation through depletion or suppression of antibody-forming plasma cells or their precursor B cells
The B lymphocytes function as antigen-presenting cells (APC) to T cells; on the other hand, the process of B-cell activation is dependent on T lymphocytes. Antibody response to protein antigens requires CD4+ helper T lymphocytes that recognize the antigen [21]. Moreover, T cell-mediated acute rejection often accompanies AMR in the biopsy. Therefore, targeting T lymphocytes has become a part of the therapeutic protocols in treatment of acute AMR. Different centers use intravenous methylprednisolone or thymoglobulin for this purpose. Although the latter may have additional effects on AMR effector cells, as discussed later. In the following section, we will concentrate on more specific treatment modalities.
Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIG) has been used for treatment of various autoimmune diseases [124]. A number of different mechanisms have been suggested for its action. Anti-idiotypic activity with neutralization of the noxious antibodies, suppression and neutralization of cytokines and activated complement components, blockade of leukocyte adhesion molecules, upregulation of inhibitory Fcγ receptor IIB, blockade of FcRn receptors and hence shortening the half-life of targeted antibodies, and blockade of activating Fcγ receptor, immunomodulation by sialylated IgG, which also promotes B-cell apoptosis, are among the proposed properties of IVIG [124–132]. IVIG effects extend beyond the half-life and presence of IVIG in circulation, attributed to its ability to modulate cell-mediated immunity through Fc receptors [133]. The different IVIG preparations differ from each other in concentration of Ig, type and content of sugar, sodium concentration, and osmotic load [123]. In addition, the anti-HLA antibody inhibitory effect of these preparations may also vary [134]. Adverse effects of IVIG occur in less than 5 % of the patients. The serious adverse reaction include aseptic meningitis, anaphylactic reaction, hypercoagulability resulting in deep venous thrombosis, pulmonary embolism, retinal vein thrombosis, stroke, myocardial infarction, clotting of dialysis access, and acute kidney injury.
For treatment of acute AMR, IVIG has been generally used with two different dosing regimens; high dose (2 g/kg), administered as a single or in split doses, and low dose (100 mg/kg), which is administered along with plasmapheresis, after each exchange treatment. The efficacy of high-dose IVIG in treatment of acute AMR was first reported by Jordan et al. [135]. They described their experience in ten patients with severe rejection, four with high levels of DSA. All the rejection episodes were reversed, and DSA titers rapidly dropped. This group have reported efficacy of high-dose IVIG in desensitization of high-PRA recipients or those with positive crossmatch [115, 136–138].
Plasmapheresis
Removing the DSA from circulation is an important component of treatment of acute AMR. Plasmapheresis provides the fastest and most effective method of achieving this goal. The modalities include removing the whole plasma and replacing the equivalent volume with albumin or fresh frozen plasma (plasma exchange) or selective removal of Ig, using double-filtration plasmapheresis [26, 139–141]. The alternative method is immunoadsorption using staphylococcal protein A column or sheep anti-IgG antibody column to remove IgG component of the plasma [142–147]. Plasma exchange is currently the only modality available in the United States. Considering the volume of distribution and rebound of the antibody, several 1–1.5 volume exchanges are required for reducing the antibody titer. Generally, one immunoadsorption treatment is equivalent to three plasma exchanges regarding the antibody removal efficacy.
The major side effects of plasma exchange include coagulopathy and bleeding diathesis, volume contraction, and potential transmission of pathogens.
Since plasmapheresis only removes the circulating DSA and does not affect the antibody-producing plasma cells or their precursor B cells, reappearance of the DSA even after several treatments is expected. Therefore, this modality is used in combination with other agents that decrease antibody production. In two small case series, combination of plasma exchange or immunoadsorption and rescue immunosuppression with tacrolimus (TAC) and MMF resulted in excellent outcome in patients with AMR [116, 148]. Abraham et al. reported their experience in 20 patients with acute AMR early posttransplant [149]. Eighteen patients with viable grafts received plasma exchanges in conjunction with replacement of azathioprine with MMF and intensification of cyclosporine with or without conversion to TAC. They reported reversal of AMR in 78 % of the cases. The responders had received more intense plasmapheresis (median of 0.9 exchanges/patient/day) compared with nonresponders (0.5 exchanges/patient/day, P = 0.0053).
On the other hand, Shah et al. reported their experience using plasmapheresis in conjunction with thymoglobulin (0.75 mg/kg/day 5–10 days) in treatment of acute AMR in seven renal transplant recipients [118]. There was one graft loss; however, the other six patients had 1-year graft survival similar to recipients without AMR. This group has shown a concentration-dependent apoptosis in plasma cells and naïve or stimulated B cells in vivo after exposure to thymoglobulin [118, 150]. This effect also has been demonstrated with myeloma cells [151].
Majority of the centers, including ours, currently use plasmapheresis in combination with low-dose IVIG after each treatment, either for desensitization or for treatment of acute AMR. With this method, removal of the circulating DSA is accompanied with supposedly antibody/complement neutralization and its reduced production. Montgomery et al. first presented the efficacy of this combination therapy in reversal of acute AMR in seven cases, three as rescue therapy and four in the setting of desensitized positive crossmatch transplants [119]. Rocha et al. treated 14 patients with early acute AMR with plasmapheresis/IVIG and reported 1-year graft survival (81 %) similar to patients treated for ACR (84 %) [117]. In another study of nine patients with acute AMR, eight responded to combination therapy with 1-year serum creatinine of 1.8 mg/dL [152]. Lehrich et al. reported their experience with this method in 22 patients and showed a significant improvement in graft survival compared to historical controls [153]. We recently reviewed the results of plasmapheresis/IVIG therapy in 47 patients with strict diagnosis of acute AMR in our center (unpublished data). The half-life of the grafts after diagnosis was 26 months, and graft survival by 4 years was only 33 %. Presence of acute ACR in the index biopsy was a poor prognostic factor.
Rituximab
CD20 is an activated-glycosylated transmembrane phosphoprotein expressed on the majority of stages of B-cell development; it is present from late pro-B cells through memory cells, but not on either early pro-B cells or plasmablasts and plasma cells [154]. It is suspected that it acts as a calcium channel in the cell membrane. Therefore, it has become a target for therapy in B-cell lymphomas. Rituximab, a chimeric murine/human monoclonal antibody, is approved in the United States for this indication. This antibody after binding to CD20 molecule on the surface of B cells leads to elimination of the cell through three proposed mechanisms, complement-dependent cytotoxicity, antibody-dependent cellular cytotoxicity (ADCC), and stimulation of apoptosis [155]. B-cell depletion may also impair the expansion of effector T cells after antigen exposure and increase the regulatory T cells [156]. These properties have made rituximab an attractive choice for including in the acute AMR treatment regimen. The dose used in treatment of lymphoma is usually weekly infusion of 375 mg/m2 for 4 consecutive weeks. However, in patients with rheumatoid arthritis, two doses of 1 g 2 weeks apart have been used [157]. Although a lower dose of 500 mg has been reported to be effective in salvage therapy for acute AMR [158]. Rituximab causes a profound and prolonged depletion of B cells in the periphery and secondary lymphoid organs. In a phase one dose escalation trial of rituximab in dialysis patients, Vieira et al. showed similarly extensive depletion of CD19+ cells after a single dose of 50, 150, and 375 mg/m2. This severe depletion persisted until about 3 months later, and by 1 year only partial recovery was observed [159]. Genberg et al. studied 43 kidney transplant recipients who received a single dose of rituximab, 91 % had complete depletion of the B lymphocytes, and in 78 % of the patients with late samples, CD19+ cells remained <5 cells/mL for as long as 14–16 months [160]. They also demonstrated significant reduction of these cells in lymph nodes and the allografts. Zarkhin et al. examined the B-cell phenotype before and after treatment of rejection with rituximab and showed that in patients with rejection naïve/memory B-cell ratio is reduced compared to stable patients, and after rituximab therapy this ratio recovers back to the level seen in stable recipients by 1 year [161].
Potential acute adverse effects of rituximab include acute bronchospasm, angioedema, respiratory distress, cardiogenic shock, and anaphylaxis, particularly in patient with high pretherapy leukocyte counts [162]. It may cause reactivation of hepatitis B or C, CMV, and mycobacterium tuberculosis, and cases of Progressive multifocal leukoencephalopathy (PML) caused by JC virus have been reported.
Becker et al. used a single dose of rituximab as rescue therapy in 27 sensitized patients with refractory acute rejection (no testing for DSA or C4d) and observed significant improvement in graft function and 85 % graft survival at 2 years [163]. Faguer et al. reported their experience in eight patients with acute AMR who received 4 weekly doses of rituximab in addition to plasma exchange [164]. Graft survival after a median of 10 months was 75 % and mean serum creatinine was 1.7 mg/dL. Kaposztas et al. retrospectively compared 26 patients who received plasmapheresis and rituximab for treatment of acute AMR with 28 who only received plasmapheresis [165]. Although patient survival was similar between the two groups, 2-year graft survival was significantly better with rituximab (90 % vs. 60 %, P = 0.005). Jordan et al. have used a combination of high-dose IVIG and rituximab for desensitizing highly sensitized renal transplant candidates [166]. In 76 patients who were transplanted after receiving this regimen, 22 (29 %) experienced acute AMR early posttransplant and were treated with the same combination. Six cases lost their grafts due to AMR, with 2-year graft survival of 84 % [167]. Mulley et al. reported their experience with a single low-fixed dose of 500 mg rituximab in seven patients with acute AMR unresponsive to plasmapheresis/IVIG [158]. After a mean follow-up of 21 months, graft survival was 100 % and serum creatinine levels were significantly lower than the time of diagnosis. Lefaucheur et al. compared the efficacy of 2 weekly doses of rituximab 375 mg/m2 after plasmapheresis and high-dose IVIG in treatment of acute AMR in 12 patients to historical controls treated only with high-dose IVIG [168]. Graft survival at 3 years was 91.7 % in the rituximab group and 50 % in the control group (P = 0.02), and DSA levels were significantly lower 3 months post-rejection in the former group.
Bortezomib
As discussed previously, plasma cells that are the producers of the antibodies are not affected by rituximab and the other modalities discussed. Bortezomib is a proteasome inhibitor that is widely used for treatment of multiple myeloma. By reversibly inhibiting 26S proteasome with high affinity and specificity, it disrupts degradation of ubiquitinylated, abnormal, and misfolded proteins, leading to cell apoptosis [169]. Studies, in vitro and in vivo, have shown that this drug has a propensity to cause apoptosis in CD138+ plasma cells [141, 170, 171]. In addition, bortezomib may cause apoptosis of TH cells and reduction in B cells [172]. The regimen for treatment of multiple myeloma includes cycles of four doses at 1.3 mg/m2 on days 1, 4, 7, and 11.
Idica et al. demonstrated the ability of this drug in reversal of DSA in kidney transplant patients [173]. They showed that in 10 out of 13 patients (77 %), DSA disappeared and in the other three the titer was significantly reduced. This group then reported the use of bortezomib in six patients experiencing eight episodes of mixed AMR and ACR [174]. In all cases, the rejection was reversed and graft function improved, and there was a profound and prolonged reduction in DSA. The major side effects including thrombocytopenia, gastrointestinal toxicity, and paresthesias were transient. Perry et al. compared the activity of four agents, rituximab, thymoglobulin, IVIG, and bortezomib, on plasma cells and antibody production [170]. They showed that in vitro only bortezomib caused plasma cell apoptosis and blocked anti-HLA antibody secretion. In two patients treated with this agent for acute AMR, they demonstrated a transient decrease in plasma cells in the bone marrow and a significant reduction in DSA. Trivedi et al. used this drug in conjunction with intravenous methylprednisolone and plasmapheresis among 11 patients with anti-HLA antibodies (eight with DSA), in the absence of acute AMR, and observed successful reduction in antibody MFI to less than 1,000 in 9 within a median of 22 days after initiation of treatment [175]. The largest experience has been reported by Flechner et al. in a series of 20 patients with AMR diagnosed 19.8 months (mean) posttransplant who received a single dose of bortezomib in addition of plasmapheresis and high-dose IVIG [176]. After a mean follow-up of 9.8 months, graft survival was 85 %, estimated glomerular filtration rate was 41.9 ± 16.8 mL/min, and 59 % had significant proteinuria.
Sureshkumar et al. described disappearance of DSA and improved renal function in two patients with early acute AMR after addition of bortezomib to plasmapheresis and IVIG [177]. We observed disappearance of DSA in a patient with early AMR who was treated with plasmapheresis, IVIG, and rituximab after receiving bortezomib. But in two cases with late AMR, this agent was not effective in achieving significant reduction in dominant DSAs [178].
Complement Blockers
As discussed previously, complement activation is a major mechanism of DSA-induced graft injury, and C5 component plays a central role by releasing C5a and formation of MAC with other components (C6-9). Therefore, C5 seems to be a good target for abrogating DSA effect on the organ. Eculizumab is a humanized monoclonal antibody directed against complement protein C5, inhibiting conversion of C5 to C5b that is approved for use in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome [179]. Locke et al. presented a single case of severe AMR in a young recipient with previous history of failed renal allograft and autologous bone marrow transplant for Hodgkin’s lymphoma, who received a kidney from his mother with positive crossmatch [180]. Despite plasmapheresis and IVIG, he remained anuric. After receiving eculizumab, rituximab, and IVIG, his crossmatch became negative and ultimately his creatinine improved to 1.1 mg/dL. However, 4 months after transplant, the patient died of pulmonary hemorrhage due to veno-occlusive disease. This group later reported a highly sensitized recipient who had received bortezomib, plasmapheresis, and rituximab preconditioning and underwent a flow-positive crossmatch transplantation and experienced early AMR [181]. Eculizumab was started and the rejection was reversed, with normal serum creatinine at 6 months and negative flow crossmatch. Stegall et al. examined the efficacy of eculizumab at the time of transplant and postoperatively up to 13 weeks in living donor kidney recipients with positive crossmatch who received thymoglobulin induction with or without plasma exchange [39]. The incidence of acute AMR in the first 3 months posttransplant was 7.7 % (2/26) compared with 41.2 % in the historical control (P = 0.003). Similar numbers for development of transplant glomerulopathy at 1 year were 6.7 % (1/15) vs. 35.7 % (15/42) (P = 0.044).
Patients who are considered for this therapy should receive prior vaccination against meningococcus and pneumococcus.
The other complement target that has been studied in animal models is the C1 complex. Use of recombinant C1-inhibitor that disengages C1r and C1s in primate kidney transplant model has been effective in preventing acute AMR in sensitized animals [182]. This protein also inhibits the lectin pathway, kallikrein, and the coagulation components, factors XIIa and XIa. The potential advantage of this protein compared to eculizumab is that it does not affect the alternate pathway of complement activation, hence decreases the risk of certain infections [29].
Splenectomy
Splenectomy that has been mostly used in desensitization for ABO-incompatible transplantation has been used as rescue therapy in cases with persistent acute AMR. By substantial and immediate reduction in B-cell and plasma cell mass, splenectomy could lead to a reduction in DSA production to an extent that other adjunctive therapies could control the AMR [183] Locke et al. reported five patients experiencing severe AMR after positive crossmatch transplantation, unresponsive to plasmapheresis/IVIG and rituximab who underwent splenectomy and experienced reversal of rejection [183, 184]. Kaplan et al. reported four cases with acute AMR unresponsive to plasmapheresis/IVIG and rituximab or alemtuzumab, in the setting of ABO-incompatible transplantation in two and positive crossmatch in one, who underwent splenectomy and showed rapid decline in serum creatinine and reduction in antibody levels [185]. Others have also reported successful results after splenectomy for refractory acute AMR, unresponsive to standard therapy [186, 187].
Treatment Summary and Recommendation
Optimal treatment of acute AMR remains unclear. The treatment modalities reviewed have been used by extrapolation from other clinical conditions and supportive experimental data. The clinical experience with these options individually or in combination has been reported only in small cohorts and by retrospective comparisons. Randomized clinical trials have not been feasible. Therefore, adopting a treatment strategy can only be based on available literature and local experience in the patient population treated in the center. The following regimen is a reasonable approach to treatment of acute AMR based on the presented data and our own experience.
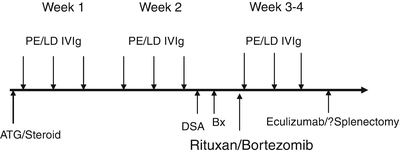
With biopsy-proven acute AMR, in conjunction with documented DSA and graft dysfunction, treatment can be started with steroid pulse and initiation of alternate day plasmapheresis (plasma exchange or immunoadsorption) with low-dose IVIG after each treatment. In cases with concomitant moderate to severe ACR, thymoglobulin can replace steroid pulse. If the graft dysfunction and histological evidence of AMR persist after 2 weeks of treatment in addition to continuation of plasmapheresis/IVIG, a single dose of rituximab or a cycle of bortezomib could be added. Failure to control AMR may warrant addition of eculizumab and in extreme cases splenectomy.
Chronic Antibody-Mediated Rejection
Introduction
Chronic AMR is a clinicopathological manifestation of chronic active graft injury as a result of donor-directed antibody. The dominant histological manifestation of this process is transplant glomerulopathy (TG). It is typically accompanied by various degrees of IFTA, multilayering of the PTC basement membrane, or arterial fibrous intimal thickening [110]. In the presence of these histological findings, presence of circulating DSA and footprint of its action in the tissue, i.e., PTC C4d, are required to label this syndrome as chronic AMR. However, an increasing number of studies point to cases with negative C4d in the biopsy [59, 71, 100, 188]. In this section, we will focus our discussion on TG as the main manifestation of chronic AMR.
Epidemiology
TG is characterized by double contours in more than 10 % of glomerular capillary loops. The reported incidence and prevalence of this entity varies. Using only for-cause biopsies, the estimated prevalence of TG was approximately 3 % of the patients at a median of 6 years posttransplant [189, 190]. Gloor et al. examined both protocol and for-cause biopsies and observed that 9.5 % of the patients with a mean follow-up of 41 ± 17 months had TG at a median of 21 ± 14 months (range 4–61) after transplant [191]. Interestingly, almost half of the patients were diagnosed based on the protocol biopsies in well-functioning grafts, this suggests that the clinical presentation of TG can lag behind the histological changes, and using only indication biopsies can underestimate its true prevalence. In this study, the overall cumulative incidence of TG increased from 4 % at 1 year to 20 % at 5 years, whereas the respective incidences in surveillance biopsies were 2.8 and 11.5 % [191].
TG is strongly and independently associated with poor graft survival [191]. In the Mayo Clinic experience, graft survival was 62 % with TG compared with 95 % without TG (P < 0.0001). Mean graft survival after the diagnosis of TG was 43 ± 7 months. The variables associated with reduced graft survival in TG cases included graft function and proteinuria at diagnosis, the severity of PTC-itis, total interstitial inflammation, the severity of interstitial fibrosis, and cg score [190, 192–194]. PTC basement membrane multilayering (PTCBMML) and arterial hyalinosis were also significant predictors of graft survival in TG [192]. The presence of any grade TG and chronic allograft vasculopathy is poor prognostic factor [195].
The association of PTC C4d staining and graft outcome in TG is controversial. Some studies have shown no association [190, 194], whereas others have documented a negative impact of late C4d positivity on graft survival [193]. C4d positivity was associated with a more rapid rate in functional decline [192]. In another study, C4d alone did not independently predict graft loss but the combined presence of C4d positivity, TG, and serum creatinine of >2.3 mg/dL at biopsy was a poor prognostic factor [196]. In the report from the Edmonton group, the cumulative death-censored graft survival was 40 % in TG with anti-HLA antibodies/C4d compared with 35 % in TG without anti-HLA antibodies/C4d at 15-year posttransplant, with no significant statistical difference [189]. We examined the outcome in 145 patients with TG at our center (unpublished data). Graft survival at 66 months of diagnosis was only 10 %. Diffuse PTC C4d by IHC was predictive of worse graft survival.
Pathogenesis
The role of donor-directed alloantibody in pathogenesis of graft in TG injury has been clearly demonstrated. Different animal models have been used to demonstrate its pathogenicity and the time course of events in TG. In a T-cell-deficient rat model, Koch et al. showed that injecting a monoclonal antibody against a mismatched nonclassical major histocompatibility complex (MHC), Ib antigen caused massive proteinuria and severe glomerulopathy [197]. de Heer et al. found that Lewis rats carrying a Fisher 344 renal allograft with TG produce antibodies against one or more novel antigen(s) of donor glomerular and tubular basement membranes [198]. In a cynomolgus monkey model, Colvin et al. described the natural history of chronic AMR [22, 199]. The authors proposed that the process starts with development of DSA followed by appearance of C4d in the biopsy leading to histological changes of TG and later graft dysfunction.
In the cohort reported from Mayo Clinic, 76 % of TG cases had detectable anti-HLA antibodies. Of those, 71 % were against class I, 90 % were against class II, and 61 % were against both [191]. TG cases with detectable HLA antibodies were more likely to be women, younger, and recipients of second transplants [193]. The incidence of TG was significantly higher among patients with anti-class II antibodies than among patients with anti-class I antibodies [193]. Examining biopsies for cause, we observed that DSA alone or in combination with C4d positivity was predictive of TG (OR: 5.8, P = 0.02, 95% CI: 1.3–25.4 for C4d−/DSA+ and OR: 17.9, P < 0.001, 95% CI: 4.9–65.2 for C4d+/DSA+ group). Furthermore, TG was associated only with class II DSA, either alone or with class I (OR: 8.3, P < 0.001, 95% CI: 2.8–24.8 for class II only and OR: 14.1, P < 0.001, 95% CI: 4.9–40.0 for class I/II DSA). In patients with DSA and positive crossmatch prior to transplant, the 1-year incidence of TG is increased to 22 % [200]. Sis et al. reported that 70 % of the patients with TG had detectable anti-HLA antibodies, 61 % class I, 91 % class II, and 52 % both [189]. DSA was present in 85 % of those with positive antibodies, 50 % class I, 86 % II, and 36 % both. The risk of TG is even significantly higher in patients with non-DSA-HLA class II antibody compared to those with no antibodies [191]. This could be due to limitations in detecting HLA-DSA with current techniques, association with non-HLA-DSA that are not assayed, cross-reactivity due to shared epitopes, or low levels of DSA, all absorbed in the graft [22, 90, 103, 200–202]. The risk of TG is not different between patients with antibodies against DR or DQ antigens [22, 193, 201].
The following steps in pathogenesis of TG are plausible, supported by available data. The initial phase is binding of the DSA, particularly class II antibodies, to allograft endothelial cells (EC). Through complement-dependent and complement-independent mechanisms, as described previously, the antibodies cause phenotypic changes in EC, presenting as cell swelling in the initial phases. Inflow of inflammatory cells especially monocytes ensue [99, 203]. With ongoing EC injury, diminished patency of the capillary loops and progression to reduplication of the glomerular basement membrane occurs. It is unclear how DSA-induced EC injury results in this reduplication and what role the recruited inflammatory cells play. Over time, EC injury may evolve into pancapillary multilayering of basement membranes and obliteration of PTC, leading to IFTA, likely secondary to ischemia [204]. Although it seems that TG develops as a result of ongoing endothelial cell activation and injury by the humoral response, it has been hypothesized that in some cases, it may occur following a period of accommodation, broken by an acute injury [103].
PTC deposition of complement-split product C4d is present in a subgroup of TG cases, supporting the participation of the classical pathway of complement activation in the pathogenesis of chronic AMR [205]. However, some TG cases have negative C4d staining, suggesting involvement of complement-independent mechanisms. Fahim et al. have reported a predominance of monocytes compared with T cells in PTC and glomeruli in C4d-positive biopsies [96]. Our group has shown the strong association of glomerular CD 68-positive cells (macrophages) with TG and as the only independent predictor of graft survival [99]. One can hypothesize that after initial stages of injury that are DSA-dependent, the subsequent stages of establishment and progression of TG and nephron mass loss are mediated by the recruited inflammatory cells, particularly monocyte/macrophages. The presence of cellular infiltrates in glomeruli (glomerulitis) is currently not considered as one of the diagnostic criteria for TG by the Banff classification, but this requires revisiting.
Proinflammatory cytokines including interleukin (IL)-1β, IL-6, and TNF-α, secreted by activated monocytes in peripheral blood, have been associated with TG, with ex vivo data supporting the role of regulatory T cells in downregulating these cytokines [91].
Elster et al. using low-density arrays identified increased expression of 57 individual genes in TG compared with stable function biopsies [206]. The Bayesian analysis identified association between ICAM-1, IL-10, CCL3, CD86, VCAM-1, MMP-9, MMP-7, and LAMC2 and allograft pathology.
Clinical Manifestation
The clinical manifestation of TG is generally insidious and nonspecific, consisting of proteinuria, unexplained loss of allograft function, and hypertension. Proteinuria is typically minor, but it can be in the nephrotic range. TG biopsies with proteinuria had more severe glomerular double contours (cg score) and increase in mesangial matrix (mm score) in comparison to TG without proteinuria [189].
At the time of 1-year protocol biopsies, patients with TG, when compared to non-TG cases, had no difference in serum creatinine concentration or estimated GFR but had a marginal increase in proteinuria [191]. Although poorer graft function and increased proteinuria were associated with the severity of TG classified by cg scores, the association was not statistically significant [191].
A great majority of cases with TG are diagnosed after the first year posttransplant. However, TG can be seen as early as 2–3 months after transplantation.
Histology
According to Banff criteria, the morphologic characteristics of chronic AMR include glomerular double contours in more than 10 % of glomerular capillary loops and/or peritubular basement membrane multilayering and/or interstitial fibrosis/tubular atrophy and/or fibrous intimal thickening in arteries [22] (Table 26.2, Fig. 26.3).
Table 26.2
Banff criteria for diagnosis of chronic AMR
Morphological features |
• Transplant glomerulopathy (double contours) and/or |
• PTC BM multilayering and/or |
• IF/TA and/or
Related posts: Live Donor Transplantation
Psychosocial Issues in Renal Transplantation
Drug Interactions in Solid Organ Transplant Recipients Live Donor Transplantation
Psychosocial Issues in Renal Transplantation
Drug Interactions in Solid Organ Transplant Recipients
 Prevention and Management of Infectious Complications in Kidney Transplant Recipients Prevention and Management of Infectious Complications in Kidney Transplant Recipients
 Urologic Issues in the Renal Transplant Patient Urologic Issues in the Renal Transplant Patient
 Immunological Assessment of the Transplant Patient Immunological Assessment of the Transplant Patient
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|