Antibiotic- and Immunosuppression-Related Renal Failure
Antibiotic- and Immunosuppression-Related Renal Failure
Marc E. De Broe
AMINOGLYCOSIDE ANTIBIOTICS
Nephrotoxic injury is a common complication of aminoglycoside antibiotic therapy. Studies that have used well-defined measures of nephrotoxicity indicate an incidence rate of 7% to 36%.1,2,3,4,5,6,7,8,9 This variability reflects differences with respect to the nephrotoxicity potentials of aminoglycoside antibiotics in clinical use as well as differences among patients receiving these drugs. A survey of clinical studies published between 1975 and 1982 revealed that the average incidence of nephrotoxicity caused by specific aminoglycoside antibiotics was gentamicin, 14%; tobramycin, 12.9%; amikacin, 9.4%; and netilmicin, 8.9%.10 In critically ill patients, the incidence of aminoglycoside nephrotoxicity may rise twofold.11
Clinical Aspects
The clinical expression of aminoglycoside nephrotoxicity has been well described.12,13,14,15,16 Lopez-Novoa and colleagues wrote a comprehensive review on the mechanisms of aminoglycoside nephrotoxicity.17 The earliest and most common expression of aminoglycoside renal tubular cell alterations is increased urinary excretion of low molecular weight proteins18,19 and of lysosomal and brush-border membrane enzymes.18,19,20,21 These changes may be detected within 24 hours of initiating drug therapy, and the frequency and magnitude of these changes increase as a function of dose and duration of therapy. Unfortunately, these changes do not predict which patients will progress to acute renal failure (ARF). This probably reflects the fact that several mechanisms underlie the expression of the enzymuria and proteinuria.13 With repeated dosing, the amount of enzymes and low molecular weight proteins excreted in the urine may increase quite sharply, which may signify the onset of proximal tubular cell necrosis.13
Nonoliguric renal failure is a common expression of aminoglycoside nephrotoxicity22 and may reflect a direct inhibitory effect on solute transport along the thick ascending limb of Henle’s loop23 or possibly tubulointerstitial cell injury,24 which results in impaired ability to maintain a hypertonic medullary interstitium. Inhibition of adenylate cyclase may also contribute to the polyuria.25 Neither mechanism, however, adequately explains the maintenance of normal to high urine output, even in the face of severe depression of whole kidney glomerular filtration rate (GFR). The slow evolution of ARF, which has been attributed to a variable susceptibility of renal proximal tubular cells to aminoglycoside toxicity,12,26 may allow for the development of maximal compensatory adaptation by residual intact nephrons. In addition, micropuncture experiments27 implicate a marked depression of solute and water transport along the proximal tubule such that the large increase in the fraction of filtrate escaping reabsorption along the proximal tubule may overwhelm the reabsorptive capacity of the distal nephron and contribute to the pattern of nonoliguric renal failure. When oliguria occurs, it usually signifies the influence of one or more complicating factors (e.g., ischemia or another nephrotoxin), especially if the oliguria appears early in the course of aminoglycoside administration. Studies in animals have shown that aminoglycoside therapy sensitizes the kidney to a subsequent ischemic or nephrotoxic insult,28,29,30,31,32,33,34,35,36,37 such that the severity of the ARF is substantially greater than that predicted by the sum of the individual insults. Deterioration of other proximal tubular transport processes may occur during aminoglycoside toxicity and, in rare cases, may mimic a Fanconi-like syndrome.38 Hypokalemia and hypomagnesemia secondary to renal potassium and magnesium wasting may also appear.39,40
Depression of GFR is a relatively late manifestation of aminoglycoside nephrotoxicity. In humans, depression of GFR typically does not occur before 5 to 7 days of therapy have been completed15 unless there has been a major complicating factor such as renal ischemia. Studies in animal models of aminoglycoside nephrotoxicity have implicated activation of the renin-angiotensin system,41 reduction in the size and density of glomerular endothelial fenestrae,42,43,44 tubular obstruction,45 tubular back leak,27 and release of platelet activating factor from mesangial cells46 as pathogenic factors causing depression of GFR.
The majority of patients with aminoglycoside nephrotoxicity recover renal function clinically, although in some cases the time to recovery may be prolonged.16 Chronic renal failure is a distinctly uncommon complication of pure aminoglycoside nephrotoxicity in humans, so that when it occurs, it usually signifies the contribution of some additional factor. Animal studies indicate, however, that incomplete regeneration with interstitial fibrosis does occur,47 and the same may be true for humans.48
Morphologic Alterations
Aminoglycosides cause tubular cell necrosis that in animal models is largely confined to the proximal convoluted tubule and pars recta.49,50,51 In humans, the renal tubular site of injury is less well established,24,52 due in part to the fact that little human biopsy material has been available for study. Moreover, in human subjects, the development of ARF in conjunction with aminoglycoside administration typically occurs in association with other insults such as sepsis and renal ischemia,24,53,54 and each of these insults has been shown to interact synergistically with aminoglycoside antibiotics to magnify the severity and sites of tubular cell injury.31,32,33,34,35,36,37
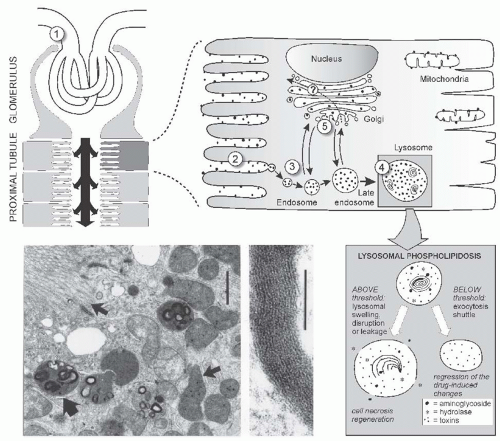
FIGURE 31.1 Above: Binding uptake and intracellular trafficking of gentamicin in renal proximal tubular cells. A: After glomerular filtration (1), gentamicin (•) is shown binding to the surface membrane (2) and being internalized by a receptor (megalin) mediated endocytic process (3). Gentamicin also enters the cell through fluid phase endocytosis. It moves through the endocytic system into late endosomes and from there into lysosomal structures (4). A small but quantifiable fraction (5%-10%) of gentamicin directly traffics from the surface membrane into the trans-Golgi network (5) and from there throughout the Golgi Apparatus. Below left: Ultrastructural appearance of proximal tubular cells after 4 days of gentamicin treatment, showing lysosomes containing dense lamellar and concentric structures (large arrow), while brush-border, mitochondria (small arrow), and peroxisomes are unaltered. Upon higher magnification the structures in lysosomes show a periodic pattern. Bar left = 1 µm, middle = 0.1 µm. Below right: Internalization and lysosomal sequestration of gentamicin. (Adapted from Verpooten GA, Tulkens PM, Molitoris BA. Aminoglycosides and vancomycin. In: De Broe ME, Porter GA, Bennett VM, Verpooten GA, eds. Clinical Nephrotoxins – Renal Injury from Drugs and Chemicals, 2nd ed. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2003:151-170.)
The earliest lesion seen by electron microscopy is an increase in the number and size of secondary lysosomes, also called cytosegrosomes or phagosomes.49,50,51 Examples of this lesion are shown in Figure 31.1. Secondary lysosomes are primary lysosomes that have coalesced with endocytic or autophagic vacuoles. Many of these lysosomes contain myeloid bodies, electron-dense lamellar structures of concentrically arranged and densely packed membranes. These lysosomal alterations probably represent autophagic vacuoles arising from sequestration of fragments of membranes and organelles damaged in the early phase of toxicity and are undergoing lysosomal processing. In experimental animals receiving single parenteral drug doses or continuous drug infusion, these changes have been observed as early as 6 to 12 hours posttreatment.56 Both the number and size of lysosomal myeloid bodies increase as a function of dose and duration of drug therapy and are accompanied by progressive expansion of the volume of the cell occupied by engorged lysosomes.50,56,57 These morphologic alterations also have been convincingly demonstrated in human kidney material.58,59 Studies in experimental animals and in cultured cells have demonstrated that the myeloid bodies are composed of membranes rich in phospholipids60,61 and form as a consequence of the lysosomal accumulation in high concentration of aminoglycosides. This lysosomal accumulation of aminoglycosides inhibits lysosomal phospholipases62,63 and possibly other lysosomal enzymes and impairs the degradation of cell membrane.61,64 Similar alterations have been induced by a variety of compounds that accumulate within the lysosomal compartment and interfere with the activity of lysosomal enzymes.65,66,67
Following lysosomal alterations, the following occurs: a decrease in the density and height of brush-border microvilli, dilation of the cisternae of rough endoplasmic reticulum, and the appearance of cytoplasmic vacuolization in tubular epithelial cells.51,56 As injury progresses, brush-border membrane fragments and extruded myeloid bodies, membrane vesicles, and cytoplasmic debris begin to be seen within tubular lumina.51,68 Later in the course of nephrotoxicity, mitochondrial swelling becomes evident, and patchy but extensive tubular epithelial cell necrosis and desquamation occur. Many tubules, both proximal and distal, are filled with eosinophilic, granular material that, by electron microscopy, is composed of cytoplasmic debris, membrane fragments, and myeloid bodies. Transmission electron microscopy of the urine reveals the presence of myeloid bodies and fragments of brush-border membranes.68,69,70
Proximal tubular cells manifest an apparent variable susceptibility to aminoglycoside toxicity evident by the appearance of cell regeneration simultaneously with ongoing cell necrosis.26,47,50,57,71 In several animal studies virtually complete recovery of renal structure and function has been observed during continued aminoglycoside administration.72,73 One explanation for these observations is that the renal tubular epithelium had acquired resistance to the nephrotoxic effects of the aminoglycoside antibiotic. Sundin and colleagues74 report that the “acquired resistance” reflects selective inhibition of aminoglycoside uptake by renal proximal tubular cells, the mechanism of which does not involve a reduction in the membrane content of phosphatidylinositol or megalin. In animal models, cell regeneration can be detected by [3H]thymidine incorporation into DNA after only 4 days of low-dose aminoglycoside administration and before cell necrosis is evident by light microscopy.57,74 The magnitude of DNA labeling correlates with the dose and duration of drug administration.75 Of particular interest is the observation that quantitatively similar labeling is observed in renal cortical interstitial cells as in tubular epithelial cells.57,75,76 This finding raises the question of the role of these interstitial cells in the pathogenesis of aminoglycoside toxicity. Eventually most areas of the affected kidney regain normal architecture and function, but residual scarring containing collections of collapsed, atrophic tubules may occur focally in the cortex.47,48,51 In animal models of aminoglycoside nephrotoxicity, the degree of tubular cell necrosis correlates reasonably well with the decline in renal excretory function. A similar correlation is lacking in human material.24,52,58
Pathogenesis
The pathogenesis of aminoglycoside nephrotoxicity is intimately linked to the renal pharmacology of these drugs.77,78,79,80 Aminoglycoside antibiotics are organic polycations with a net cationic charge that, at pH 7.4, ranges from +4.47 in the case of neomycin to +2.39 for amikacin. Because these compounds are highly hydrophilic, they are poorly absorbed across the intestinal tract and therefore must be given parenterally. They are distributed in a volume slightly greater than extracellular volume and are eliminated from the body without metabolic transformation. The route of elimination is almost exclusively by the kidneys, and the principal mechanism of excretion is glomerular filtration. Of toxicologic significance is the fact that small amounts of aminoglycoside antibiotics are selectively transported into proximal tubular cells by adsorptive endocytosis,81,82,83 which has been shown to occur across the basolateral as well as the apical membrane.83 Several lines of evidence have implicated anionic phosphatidylinositol as a membrane binding site for aminoglycosides.84,85 More recent studies also suggest a role for megalin, an endocytic receptor for cationic ligands, in the uptake of aminoglycoside antibiotics across the brush-border membrane of renal proximal tubular cells.86 Indeed, by using the specific antagonist receptor-associated protein, blocking the activity of megalin in perfused rat proximal tubules, a reduction of 20% in gentamicin clearance ensued. Nagai demonstrated similar results in rats treated with maleate, impairing the receptor-mediated uptake of megalin ligands.87 Megalin knockout mice are protected against aminoglycoside nephrotoxicity.88
Following endocytosis, the aminoglycosides are translocated into the lysosomal compartment, where they accumulate in millimolar concentrations and reside with a half-life measured in days.78 As noted, the lysosomal compartment is the site of myeloid body formation consequent to aminoglycoside-induced inhibition of lysosomal enzymes such as phospholipase, sphyngomyelase, etc. When the concentration of drug and/or the amount of lysosomal phospholipid reaches a critical threshold, an injury cascade is triggered that eventuates in irreversible cell injury with progression to necrosis.56 However, neither the sequence nor the specific mechanisms involved in the progression to cell death have been clearly established. Sandoval and colleagues report that within 15 minutes of endocytosis gentamicin traffics to the Golgi complex as well as to the lysosomal compartment of LLC-PK1 cells89,90 and rat renal proximal tubular cells.91 These observations raise the possibility that the Golgi complex may provide a pathway for the redistribution of aminoglycoside antibiotics to other intracellular compartments and thereby broaden the potential for these drugs to disrupt a variety of organellar functions. For example, the depression of protein synthesis observed early in the course of gentamicin administration may signify retrograde transport of gentamicin to the endoplasmic reticulum.91 The reason gentamicin and presumably other aminoglycoside antibiotics are transported from the endosomal compartment to the Golgi complex is not known; but, it may reflect an effect of these agents to perturb endosomal fusion92 possibly as a consequence of binding to megalin92 or to membrane-acidic phospholipids.93,94
A growing body of evidence supports the view that the pathogenesis of aminoglycoside toxicity is causally related to the capacity of these cationic drugs to bind to and perturb the function and structure of biologic membranes. Aminoglycosides have been shown to bind to anionic62,84,95,96,97,98,99,100,101,102 but not to neutral phospholipids.62,84,96,98 Among the anionic phospholipids, aminoglycosides bind most avidly to phosphatidylinositol 4,5-bisphosphate (PIP2).84,97,103,104,105 Several approaches have been used to gain insight into the molecular interaction between aminoglycosides and anionic phospholipids.84,95,98,101,102,106,107,108 All models indicate an electrostatic interaction between a protonated amino group and the anionic phosphate group. Ramsammy and Kaloyanides107 propose a model that, in addition to an electrostatic interaction between a protonated amino group and the phosphate group, also involves formation of hydrogen bonds between an amino group of gentamicin and the carbonyl groups of glycerol. This model explains aminoglycoside-induced changes in the biophysical properties of artificial membranes (i.e., an increase in the transition temperature and a decrease in glycerol permeability of phosphatidylinositol [PI]-containing liposomes).100 Both changes signify that gentamicin induces a decrease in membrane fluidity, and this finding has been confirmed in brush-border membranes as assessed by changes in the fluorescence polarization of membrane probes96 and by electron spin resonance spectroscopy.109 Aminoglycosides also have been shown to promote membrane aggregation,106,110 a process that requires neutralization of surface charge. In a comparative study of aminoglycoside-induced aggregation of PI-containing liposomes,106 it was observed that the rank order with respect to efficacy in neutralizing membrane surface charge was neomycin > gentamicin = tobramycin = netilmicin = spermine. The rank order for inducing aggregation of liposomes was neomycin > gentamicin > tobramycin > netilmicin = spermine and was identical to the rank order of these agents with respect to depressing glycerol permeability.106 This rank order also coincides precisely with the established clinical nephrotoxicity potentials of these drugs. Because depression of glycerol permeability was shown to be dependent on hydrogen bonding between one or more amino groups of the drug and carbonyl groups of the glycerol backbone,107 these data suggest that the membrane toxicity of aminoglycosides is closely linked to their potentials to engage in hydrogen bonding. Importantly, the rank order in terms of nephrotoxicity potentials does not coincide with the net cationic charge of these agents.106 This observation emphasizes that spatial orientation of charge rather than net charge is a critical determinant of toxicity.
Schacht and colleagues97,99,104,111 utilize a variety of methods to assess aminoglycoside-induced perturbations of PIP2-containing membranes as a measure of the ototoxicity potentials of these antibiotics. Increased fluorescence of 1-anilino-8-naphthalenesulfonate,99 increased permeability to carboxy fluorescein,104 and increased surface tension of monomolecular film of phosphatidylcholine (PC)/PIP2111 were shown to correlate precisely with the ototoxicity potentials of aminoglycoside antibiotics. These studies have led to the hypothesis that the ototoxicity of aminoglycosides is causally related to their binding to PIP2 and disruption of this signaling mechanism.112
The studies cited here provide the foundation for the hypothesis that the toxicity of aminoglycoside antibiotics is causally related to their capacity to interact electrostatically and by hydrogen bonding to membrane anionic phospholipids and, thereby, to perturb the biophysical properties and function of cell membranes. It is well established that these drugs interact with and perturb the function of plasma membranes,13,113,114,115,116 lysosomes,13,56,57,58,59,60,61,62,63,64,117,118,119,120,121,122 mitochondria,51,123,124,125,126 and microsomes.127,128,129 It remains unclear, however, whether toxicity results from disruption of a single critical membrane function or multiple membrane functions. It is possible that the injury cascade is triggered by the rupture of lysosomes engorged with aminoglycoside antibiotic and with myeloid bodies. The resultant release of potent acid hydrolases and high concentrations of drug into the cytoplasm might cause disruption of a number of critical intracellular processes including mitochondrial respiration,51,123,124,125,126 microsomal protein synthesis,127,128,129 intracellular signaling via the PI cascade130,131,132,133 as well as generation of hydroxyl radicals134,135,136—all of which have been observed in experimental models of aminoglycoside toxicity. However, the observation that gentamicin is transported to the Golgi complex shortly after endocytic uptake89,90 provides an alternate mechanism by which these drugs gain access to other organelles. Recently, proteomic analysis following gentamicin administration indicates energy production impairment and a mitochondrial dysfunction occurring in parallel to the onset of nephrotoxicity.137
Further insight into the pathogenesis of aminoglycoside nephrotoxicity has been gleaned from studies of interventions that modify the severity of this disorder in experimental animals. Williams and colleagues138,139,140 first reported that polyasparagine and polyaspartic acid (PAA) inhibited binding of gentamicin to rat renal brush-border membrane in vitro and when injected in vivo conferred protection against the development of aminoglycoside nephrotoxicity without inhibiting the renal cortical accumulation of drug. These findings have been confirmed and extended by three groups of investigators.141,142,143,144,145,146,147,148,149 The mechanism by which PAA protects against aminoglycoside nephrotoxicity was shown to be related to the ability of PAA, a polyanion, to form electrostatic complexes with the polycationic aminoglycoside antibiotics146,150,151 presumably within the endocytic compartment,148 thereby preventing aminoglycosides from binding to anionic phospholipids, from inhibiting lysosomal phospholipase degradation of phospholipid, from forming lysosomal myeloid bodies, and from disrupting the PI cascade.150 Additional support for this theory is provided by the observation that PAA prevented gentamicin from depressing glycerol permeability or aggregating PI-containing liposomes,150 effects previously shown to be dependent on gentamicin binding electrostatically and by hydrogen bonding to PI.100,107 Subsequently, other compounds capable of forming electrostatic complexes with aminoglycosides have been reported to protect against nephrotoxicity.152,153,154,155
An analog of pentoxifylline, HWA-448, was shown to protect against gentamicin toxicity in a cell culture model.156 Similar to PAA, HWA-448 did not depress the membrane binding or cellular uptake of gentamicin. It remains unknown whether HWA-448 forms a complex with gentamicin within the endosomal compartment.
Recently, it was demonstrated that glibenclamide (a sulfonylurea) has protective effects against gentamicin-induced nephrotoxicity in rats.157 Morales et al. suggest that the pleiotropic effects of metformin can decrease gentamicin nephrotoxicity by improving mitochondrial homeostasis.158
Treatment and Prevention of Aminoglycoside Nephrotoxicity
The efficacy of PAA and other anionic compounds in preventing nephrotoxicity in humans has yet to be established. Therefore, the primary focus of treatment is prevention, and this can be accomplished by understanding and modifying, when possible, the risk factors (Table 31.1) for this complication.159,160,161 Risk factors may be categorized into those that are determined by the individual patient and not easily influenced, if at all, and those that are determined by the clinician and potentially controllable (Table 31.1).
Prominent among the risk factors peculiar to the patient and not modifiable is advanced age.159 The mechanism is probably multifactorial and includes age-related decline of renal function that, if not appreciated and corrected for, results in excessive dosing.162 Animal studies suggest that aging is associated with altered renal pharmacokinetics accompanied by increased renal cortical accumulation of drug.163 Increased susceptibility of the aging kidney to aminoglycoside toxicity has also been suggested,164 possibly on the basis of an age-related impaired capacity for cellular repair and regeneration. Male gender has been shown to carry increased risk for aminoglycoside nephrotoxicity in the rat,165 whereas female gender has been identified as a risk factor in humans.159 The reason for this difference has not been established.
TABLE 31.1 Risk Factors for Aminoglycoside Nephrotoxicity
a Concurrent with experimental nephrotoxicity data. Adapted from Verpooten GA, Tulkens PM, Molitoris BA. Aminoglycosides and vancomycin. In: De Broe ME, Porter GA, Bennett VM, Verpooten GA, eds. Clinical Nephrotoxins — Renal Injury from Drugs and Chemicals, 2nd ed. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2003:151-170.
Obesity carries increased risk for aminoglycoside nephrotoxicity that is unexplained by differences in the volume of distribution or renal clearance of drug.166 The increased risk associated with chronic liver disease159 may be related to the alterations in extracellular volume, hemodynamics, and electrolyte balance commonly observed in this disorder, all of which are known to promote renal cortical accumulation of drug.78 Preexisting chronic renal insufficiency is associated with increased risk primarily due to failure to adjust appropriately the dose of aminoglycoside for the level of impaired kidney function.167 Renal hypoperfusion from any cause carries an increased risk of aminoglycoside nephrotoxicity whether the renal ischemic insult occurs before,85 during,34 or after drug administration.32 The latter observation is particularly worthy of note because it implies that the increased risk of nephrotoxicity persists even after the drug has been discontinued. The prolonged half-life of aminoglycosides in renal cortex78 may contribute to this risk. Three components of the septic state—renal hypoperfusion, endotoxemia, and hyperthermia—have been identified as factors contributing to the heightened risk of nephrotoxicity during aminoglycoside therapy.35,36,37 Renal hypoperfusion35,85 and endotoxemia33,168
Only gold members can continue reading. Log In or Register to continue