Iain C. Macdougall, Kai-Uwe Eckardt
Anemia in Chronic Kidney Disease
Anemia is an almost universal complication of chronic kidney disease (CKD). It contributes considerably to reduced quality of life of patients with CKD and has been associated with a number of adverse clinical outcomes. Before the availability of recombinant human erythropoietin (rHuEPO, or epoetin), patients on dialysis frequently required blood transfusions, exposing them to the risks of iron overload, transmission of viral hepatitis, and HLA sensitization, which reduced the chances of successful transplantation. The advent of rHuEPO in the late 1980s changed this situation completely. The ability to correct anemia has suggested that its consequences may go beyond general fatigue and reduced physical capacity to affect a broad spectrum of physiologic functions. Thus, there is a strong rationale for managing anemia in CKD patients, and yet the optimal treatment strategies are still incompletely defined. Apart from therapy with erythropoiesis-stimulating agents (ESAs), iron replacement is essential for anemia management. It is important to note that CKD patients require target thresholds of iron parameters different from those for normal individuals to ensure optimal rates of red cell production. The costs of anemia management are considerable, and it has also become apparent that full anemia correction may cause harm; therefore a rational and careful consideration of the risks and benefits is mandatory.
Pathogenesis
Renal anemia is typically an isolated normochromic, normocytic anemia with no leukopenia or thrombocytopenia. Both red cell life span and the rate of red cell production are reduced, but the latter is more important. The normal bone marrow has considerable capacity to increase the rate of erythropoiesis, and the reduction in erythrocyte life observed in association with CKD would normally be easily compensated for. However, this erythropoietin (EPO)–induced compensatory increase in erythrocyte production is impaired in CKD. Serum EPO levels remain within the normal range and fail to show the inverse exponential relationship with blood oxygen content characteristic of other types of anemia. EPO is normally produced by interstitial fibroblasts in the renal cortex, in close proximity to tubular epithelial cells and peritubular capillaries.1,2 In addition, hepatocytes and perisinusoidal Ito cells in the liver can produce EPO (Fig. 83-1). Hepatic EPO production dominates during fetal and early postnatal life but does not compensate for the loss of renal production in adult organisms. Subtle changes in blood oxygen content induced by anemia, reduced environmental oxygen concentrations, and high altitude stimulate the secretion of EPO through a widespread system of oxygen-dependent gene expression.2–4 Central to this process is a family of hypoxia-inducible transcription factors (HIFs). The two most important members of this family, HIF-1 and HIF-2, are composed of an oxygen-regulated α subunit (HIF-1α or HIF-2α) and a constitutive β subunit. The production of HIF-1α and HIF-2α is largely independent of oxygen, but their degradation is related to cellular oxygen concentrations. Hydroxylation of specific prolyl and asparagyl residues of HIF-α, for which molecular oxygen is required as a substrate, determines HIF proteasomal destruction and inhibits its transcriptional activity. Apart from EPO, far more than 100 HIF target genes have been identified. HIF-2, rather than HIF-1, is the transcription factor primarily responsible for the regulation of EPO production.5,6
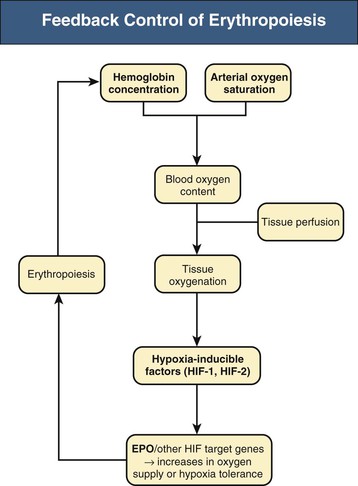
The role of renal EPO production in the pathogenesis of renal anemia is supported by the observation that anemia is particularly severe in anephric individuals. However, the mechanisms impairing renal EPO production in diseased kidneys remain poorly understood. The production capacity for EPO remains significant, even in end-stage renal disease. Thus, patients with anemia and CKD can respond with a significant increase in EPO production to an additional hypoxic stimulus.1 The main problem therefore appears to be a failure of EPO production in response to chronically reduced hemoglobin (Hb) concentrations. In line with this view, endogenous EPO production can be induced in CKD patients by pharmacologic inhibition of HIF degradation (see later discussion).
Erythropoietin is a glycoprotein hormone consisting of a 165–amino acid protein backbone and four complex, heavily sialylated carbohydrate chains.1,2 The latter are essential for the biologic activity of EPO in vivo because partially or completely deglycosylated EPO is rapidly cleared from the circulation. This is also why rHuEPO has to be manufactured in mammalian cell lines; bacteria lack the capacity to glycosylate recombinant proteins.
Erythropoietin stimulates red cell production by binding to homodimeric EPO receptors, which are primarily located on early erythroid progenitor cells, the burst-forming units erythroid (BFU-e) and the colony-forming units erythroid (CFU-e). Binding of EPO to its receptors salvages these progenitor cells and the subsequent earliest erythroblast generation from apoptosis, thereby permitting cell division and maturation into red blood cells.7 Inhibition of red cell production by uremic inhibitors of erythropoiesis may also contribute to the pathogenesis of renal anemia, although they have so far not been identified. Nevertheless, dialysis per se can improve renal anemia and the efficacy of ESAs. Moreover, the interindividual dose requirements for ESAs vary significantly among CKD patients, and the average weekly dose is much higher than estimated production rates of endogenous EPO in healthy individuals. An alternative view to the accumulation of inhibitors of erythropoiesis in CKD is that in many patients there is overlap between renal anemia and the anemia of chronic disease, which is characterized by inhibition of EPO production and EPO efficacy as well as by reduced iron availability, mediated through the effects of inflammatory cytokines.8 The hepatic release of hepcidin, the key regulator of iron metabolism, is upregulated in states of inflammation; it simultaneously blocks iron absorption from the gut and promotes iron sequestration in macrophages.9
Epidemiology and Natural History
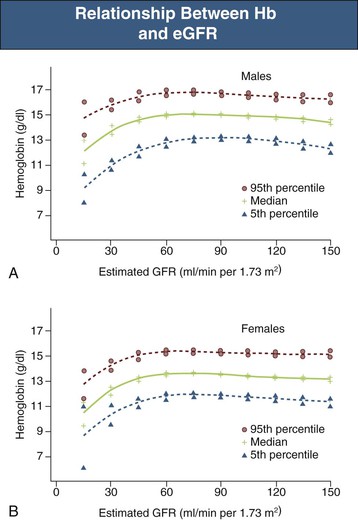
In general, there is a progressive increase in the incidence and severity of anemia with declining renal function. The reported prevalence of anemia by CKD stage varies significantly and depends to a large extent on the definition of anemia and whether study participants are selected from the general population, are at high risk for CKD, are diabetic, or are already under the care of a physician. Data from the National Health and Nutrition Examination Survey (NHANES) showed that the distribution of Hb levels starts to fall at an estimated glomerular filtration rate (eGFR) of less than 75 ml/min/1.73 m2 in men and 45 ml/min/1.73 m2 in women (Fig. 83-2).10 The prevalence of Hb values below 13 g/dl increases below a threshold eGFR of 60 ml/min/1.73 m2 in men and 45 ml/min/1.73 m2 in women in the general population. Among patients under regular care and known to have CKD, the prevalence of anemia was found to be much greater, with mean Hb levels of 12.8 ± 1.5 (CKD stages 1 and 2), 12.4 ± 1.6 (CKD stage 3), 12.0 ± 1.6 (CKD stage 4), and 10.9 ± 1.6 (CKD stage 5).11 Although anemia develops largely independently of the cause of kidney disease, there are two important exceptions. Diabetic patients develop anemia more frequently, at earlier stages of CKD, and more severely at a given level of renal impairment.12,13 Conversely, in patients with polycystic kidney disease, Hb is on average higher than in other patients with similar degrees of renal failure, and polycythemia may occasionally develop.1
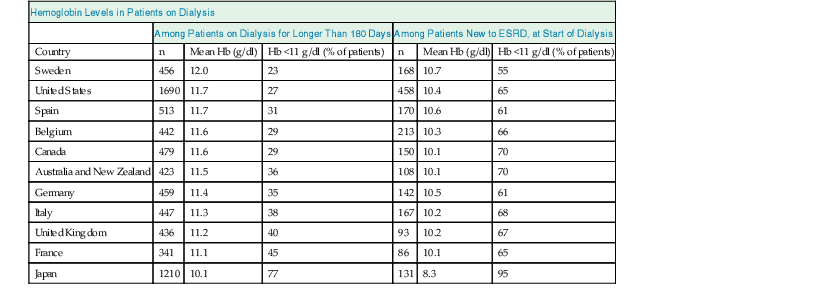
With the advent of rHuEPO and its derivatives, Hb values in patients with CKD have changed. In particular, in patients on dialysis, average Hb values have steadily increased for many years, and then declined again in view of new evidence suggesting lower target levels.12 The average Hb value, however, still varies considerably among countries, reflecting considerable variability in practice patterns (Table 83-1).13 Moreover, Hb values vary considerably among patients in the same treatment setting as well as within patients over time, mainly reflecting persistent and time-dependent changes in responsiveness (see later discussion).
Table 83-1
Mean hemoglobin (Hb) levels and percentage of patients with Hb levels below 11 g/dl who have been on dialysis therapy for more than 180 days and at the time of starting dialysis, by country.
Data are from the Dialysis Outcomes and Practice Patterns Study, Phase II (DOPPS 2), and are derived from 308 randomly selected, representative dialysis facilities. Note that there are marked differences among countries, but at least one fourth and up to three fourths of dialysis patients, and, in most countries, more than two thirds of patients starting chronic dialysis have Hb values below the recommended lower target of 11 g/dl. ESRD, End-stage renal disease.
| Hemoglobin Levels in Patients on Dialysis | ||||||
| Among Patients on Dialysis for Longer Than 180 Days | Among Patients New to ESRD, at Start of Dialysis | |||||
| Country | n | Mean Hb (g/dl) | Hb <11 g/dl (% of patients) | n | Mean Hb (g/dl) | Hb <11 g/dl (% of patients) |
| Sweden | 456 | 12.0 | 23 | 168 | 10.7 | 55 |
| United States | 1690 | 11.7 | 27 | 458 | 10.4 | 65 |
| Spain | 513 | 11.7 | 31 | 170 | 10.6 | 61 |
| Belgium | 442 | 11.6 | 29 | 213 | 10.3 | 66 |
| Canada | 479 | 11.6 | 29 | 150 | 10.1 | 70 |
| Australia and New Zealand | 423 | 11.5 | 36 | 108 | 10.1 | 70 |
| Germany | 459 | 11.4 | 35 | 142 | 10.5 | 61 |
| Italy | 447 | 11.3 | 38 | 167 | 10.2 | 68 |
| United Kingdom | 436 | 11.2 | 40 | 93 | 10.2 | 67 |
| France | 341 | 11.1 | 45 | 86 | 10.1 | 65 |
| Japan | 1210 | 10.1 | 77 | 131 | 8.3 | 95 |
(Modified from reference 13.)
Diagnosis and Differential Diagnosis
The diagnosis of anemia and the assessment of its severity are best made by measuring the Hb concentration rather than the hematocrit. Hb is a stable analyte that is measured directly in a standardized fashion, whereas the hematocrit is relatively unstable, indirectly derived by automatic analyzers, and lacking in standardization. Within-run and between-run coefficients of variation in automated analyzer measurements of Hb are one half and one third those for hematocrit, respectively.12
There is considerable variability in the Hb threshold used to define anemia. According to the most recent definition in the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, anemia should be diagnosed at Hb concentrations of less than 13.0 g/dl in men and less than 12.0 g/dl in women.14 These values represent the World Health Organization (WHO) definition of anemia. In children, age-dependent differences in the normal values have to be taken into account. Normal Hb values are increased in high-altitude residents. It is important to note that thresholds for the diagnosis of anemia and evaluation of the causes should not be interpreted as being thresholds for treatment of anemia.14
In addition to the Hb value, the evaluation of anemia in CKD patients should include a complete blood count with red blood cell indices (mean corpuscular Hb concentration [MCHC], mean corpuscular volume [MCV]), white blood cell count (including differential), and platelet count. Although renal anemia is typically normochromic and normocytic, deficiency of vitamin B12 or folate may lead to macrocytosis, whereas iron deficiency or inherited disorders of Hb formation (such as thalassemia) may produce microcytosis. Macrocytosis with leukopenia or thrombocytopenia suggests a generalized disorder of hematopoiesis caused by toxins, nutritional deficit, or myelodysplasia. Hypochromia probably reflects iron-deficient erythropoiesis. An absolute reticulocyte count, which normally ranges between 40,000 and 50,000 cells/µl of blood, is a useful marker of erythropoietic activity.
Iron status tests should be performed to assess the level of iron in tissue stores or the adequacy of iron supply for erythropoiesis. Although serum ferritin is so far the only available marker of storage iron, several tests reflect the adequacy of iron for erythropoiesis, including transferrin saturation, the percentage of hypochromic red blood cells (PHRC), the content of Hb in reticulocytes (CHr), and the MCV and MCHC. Storage time of the blood sample may elevate PHRC, and MCV and MCHC are below the normal range only after longstanding iron deficiency.
It is important to identify anemia in CKD patients because it may signify nutritional deficits, systemic illness, or other conditions that warrant attention, and even at modest degrees, anemia reflects an independent risk factor for hospitalizations, cardiovascular disease, and mortality.12 The diagnosis of renal anemia, that is, an anemia caused by CKD, requires careful judgment of the degree of anemia in relation to the degree of renal impairment and exclusion of other or additional causes. Because there is significant variability in the degree of anemia in relation to the impairment in renal function, no simple diagnostic criteria can be applied. Causes of anemia other than EPO deficiency should be considered when (1) the severity of anemia is disproportionate to the impairment of renal function, (2) there is evidence of iron deficiency, or (3) there is evidence of leukopenia or thrombocytopenia. Concomitant conditions such as sickle cell disease may exacerbate the anemia, as can drug therapy. For example, inhibitors of the renin-angiotensin system may reduce Hb levels by (1) direct effects of angiotensin II on erythroid progenitor cells,15 (2) accumulation of N-acetyl-seryl-lysyl-proline (Ac-SDKP), an endogenous inhibitor of erythropoiesis in patients treated with angiotensin-converting enzyme (ACE) inhibitors,16 and (3) reduction of endogenous EPO production, possibly because of the hemodynamic effects of angiotensin II inhibition. Myelosuppressive effects of immunosuppressants may further contribute to anemia.17 The measurement of serum EPO concentrations is usually not helpful in the diagnosis of renal anemia because there is relative rather than absolute deficiency, with a wide range of EPO concentrations for a given Hb concentration that extends far beyond the normal range of EPO levels in healthy, nonanemic individuals. Abnormalities of other laboratory parameters should be looked for, such as a low MCV or MCHC (may indicate an underlying hemoglobinopathy), a high MCV (may indicate vitamin B12 or folate deficiency), or an abnormal leukocyte or platelet count (may suggest a primary bone marrow problem, such as myeloma or myelodysplastic syndrome), and further tests should be performed as indicated to explore these potential conditions. However, when there are no such pointers to other confounding causes of anemia, and iron deficiency has been excluded, a trial with rHuEPO or its derivatives is warranted, even when the eGFR is only moderately reduced.
Clinical Manifestations
In the early clinical trials of rHuEPO performed in the late 1980s, the mean baseline Hb concentration was about 6 to 7 g/dl, and this progressively increased to about 11 or 12 g/dl after treatment. Patients subjectively felt much better, with reduced fatigue, increased energy levels, and enhanced physical capacity, and there were also objective improvements in cardiorespiratory function.18 Thus, it is now clear that many of the symptoms previously attributed to the “uremic syndrome” may have been caused by severe anemia associated with CKD (Boxes 83-1 and 83-2). Although the avoidance of blood transfusions and improvement in quality of life are obvious early changes, there are also possible effects on the cardiovascular system (see Box 83-1
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


