For most of its medical history, the kidney has been considered a tubular organ. Even after Marcello Malpighi (1628-1694) described his eponymous “corpuscles” and William Bowman (1816-1892) that of his “capsule,” the kidney continued to be viewed as a secretory tubular organ. In fact, after the milestone report of dropsical albuminuric medical cases by Richard Bright (1789-1858), which launched the study of kidney disease, the inflammatory diseases of the kidney that he described were classified in 1858 by Rudolph Virchow (1821-1902) as a nephritis principally affecting the tubules (“parenchymatous” nephritis) or the interstitium (“interstitial” nephritis). It is only after the improved resolution of microscopes and refinements in tissue processing that in 1869 “glomerular” nephritis was added to the list of nephritides by Edwin Klebs (1834-1913). Still, most diseases of the kidney continued to be considered as tubulopathies rather than glomerulopathies well into the first decades of the 20th century. It is within this context that the pathologic diagnosis of acute interstitial nephritis (AIN) was described in 1898 by William Councilman (1854-1933). In his now classic paper, based on a review of the literature and his postmortem examination of 42 cases, he reported on a nonsuppurative inflammatory interstitial lesion of the kidney occurring predominantly in patients with diphtheria (24 cases), scarlet fever (10 cases), and less frequently in those with other infectious diseases. He described the pathologic changes as
an acute inflammation of the kidney characterized by cellular and fluid exudation in the interstitial tissue, accompanied by, but not dependent on, degeneration of the epithelium; the exudation is not purulent in character, and the lesions may be both diffuse and focal
and identified the infiltrating cells as “plasma cells that had migrated from the blood vessels and multiplied locally by mitotic division.” He localized the foci of cellular infiltrates to three sites: the boundary zone of the pyramids, the subcapsular region of the cortex, and around the glomeruli. These meticulous observations and the lucid description are just as valid today as they were then, and they encapsulate the lesions of this disease of the kidneys more succinctly and thoroughly than much of what has been written since they were first reported.
1,
2Subsequent reports confirmed Councilman’s observation that nonsuppurative lesions of the renal interstitium appeared after short but variable periods from the onset of acute infections that were primarily streptococcal in origin.
3,
4 This concept of acute tubular and interstitial injury following an acute infection was so well accepted that initial reports of acute tubular necrosis (ATN), which appeared before the proper characterization of acute renal failure due to tubular necrosis during World War II, termed the new lesion
acute hematogenous interstitial nephritis and argued for the similarity of the lesions of ATN to those of AIN.
5 The difficulty in the differential diagnosis of these two variants of tubulointerstitial disease persists to this day if the kidney tissue is not examined carefully and is interpreted without benefit of the clinical history.
1,
6,
7 The introduction of antibacterials during World War II and the eradication of serious and fatal streptococcal infections resulted in a loss of interest in the entity described by Councilman, whereas attention focused on ischemic or nephrotoxic ATN as the predominant cause of acute kidney injury (AKI). Therefore, it is ironic that interest in AIN was revived in the 1960s when the very antibiotics used to treat streptococcal infections were identified as a cause of AIN.
1,
8,
9 In fact, the bulk of current reports in the literature are on drug-induced AIN, and the number of drugs implicated as causing AIN continues to increase, as does that of the variations in their clinical and laboratory manifestations.
8,
9,
10 AIN has since come to define the clinicopathologic syndrome that develops in diverse conditions, including infections, and is characterized by an acute deterioration of kidney function, the pathologic features of which remain those first described by Councilman as “cellular and fluid exudation in the interstitial tissue, accompanied by, but not dependent on, degeneration of the epithelium … and the lesions may be both diffuse and focal.”
2Although this clinicopathologic syndrome initially was referred to as
acute interstitial nephritis,2 the more inclusive
and precise descriptive terms
acute tubulointerstitial nephritis (ATIN) and
acute tubulointerstitial nephropathy are used now.
1,
11 The preferential use of
tubulointerstitial stems from the fact that although the dominant morphologic features are those evident in the interstitium, the tubules also are affected, albeit to a degree that may be difficult to detect in some cases on light microscopy. Importantly, and independent of the severity of their structural injury, the disorders of tubular function constitute a characteristic component of the disordered kidney function and differentiate this entity from other forms of AKI due to glomerular or vascular disease. In fact, tubular dysfunction is an invariable accompanying feature of the reduction in glomerular filtration rate (GFR) in ATIN and, as a rule, precedes any clinically evident decrease in GFR.
1 In addition, although the interstitial cellular infiltrates contribute significantly to the pathogenesis of the disease, there is increasing evidence that the tubules play an important role in the processing and presentation of the antigenic stimulus and the immunopathogenesis of the disease process.
12,
13,
14,
15 Although ATIN may be suspected clinically, its diagnosis depends on the presence of characteristic morphologic changes of the renal parenchyma. The adjective
acute refers to the sudden onset and rapid progression of the clinical features of this syndrome and not to its pathologic features, which are notable for mononuclear cell infiltration rather than the polymorphonuclear leukocytes characteristic of an acute inflammatory reaction.
PATHOLOGIC FEATURES
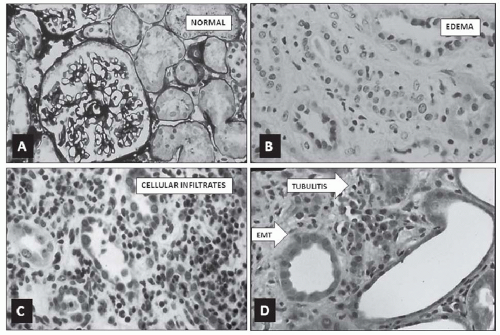
Independent of the causative agent or the clinical condition responsible for ATIN, the principal morphologic features that characterize it are an increase in interstitial volume, mainly because of edema; mononuclear cell infiltrates of varying degrees and distribution; tubular injury and degeneration of differing severity, which in general are localized to the sites of the greatest cellular infiltrates; and the transformation of tubular epithelial cells into migratory fibroblasts that cross the basement membrane (
Fig. 35.1). The glomeruli are generally spared, but may show some degree of periglomerular infiltrates and ischemic change except in ATIN associated with nephrotic syndrome, where an effacement of the podocyte foot processes is seen on electron microscopy. Although the extent and severity of each of the tubulointerstitial lesions show some correlation with the level of reduction in GFR, the closest correlation appears to be with the infiltrative process. The interstitial infiltrative lesions may be
diffuse but usually are patchy in distribution and are most evident in the cortex and corticomedullary junction, and less commonly in the outer medulla of the kidney. When the infiltrating cells are meager, they show a focal peritubular distribution that is most evident in the corticomedullary region; when profuse, they are more prominent at certain foci but literally obscure the normal appearance of the tubules and extend to the periglomerular space (
Fig. 35.1C). Diffuse infiltrative lesions are associated with higher levels of serum creatinine and a poorer prognosis for recovery than patchy infiltrative ones.
1,
8,
9The infiltrating cells are composed mostly of mononuclear cells, lymphocytes, and plasma cells, and rarely, polymorphonuclear cells and macrophages. The prognosis appears to be less favorable when 1% to 6% of the interstitial infiltrating cells are composed of neutrophilic granulocytes.
1,
9 By the same token, an increase in the number of macrophages (usually <10%) and the presence of granulomatous reactions are associated with a prolonged course of AKI and varying degrees of residual impairment of kidney function.
1,
9,
16 When present, eosinophils are sparse and constitute only a small component of the infiltrating cells, except in occasional cases of antibiotic-induced ATIN, where they may be marked. The presence of infiltrating eosinophils shows no relation to the increased eosinophilia or the presence of urinary eosinophils that also occur in drug-induced ATIN. A role for a specific eosinophil chemoattractant cytokine, eotaxin, which is expressed in the kidney, has been proposed as a mechanism for the tissue eosinophilia of ATIN.
17,
18An immunologic characterization and an analysis of the interstitial cellular infiltrates reveals that most (up to 80%) of the mononuclear cells are activated T lymphocytes. B lymphocytes also are present but constitute a much smaller portion of the cellular infiltrates, being highest in cases due to nonsteroidal anti-inflammatory drugs (NSAIDs).
1,
8 The infiltrating T cells are predominantly CD4+ and CD8+ cells. In most cases, the CD4 variety predominates over the CD8 variety.
1,
19 Natural killer lymphocytes are rare; when present, they constitute only a small proportion of the infiltrating cells.
18 A slight prevalence (just over 50%) of the CD8+ subset has been reported in cases of ATIN associated with massive proteinuria after exposure to NSAIDs.
8,
9 The relevance of this observation to massive proteinuria as a feature of ATIN is unknown.
1,
19 Convincing evidence has been advanced that the infiltrating cells are antigenically activated and immunologically engaged in the pathogenesis of the lesions. However, no diagnostic pattern of markers or cell types has come to be identified with the lesions associated with any specific form of ATIN.
1,
20Variable degrees of tubular injury usually are present, but tubular atrophy is absent. The tubular lesions are most evident at the site of the greatest concentration of infiltrating cells. They usually are focal and are more severe in the proximity of infiltrating cells. A distinguishing lesion that results from these localized peritubular infiltrates is the disruption of the tubular basement membrane (TBM) and its epithelial cell lining, so-called “tubulitis,” which is characteristic of ATIN (
Fig. 35.1D). Granulomatous reactions, multinucleated epithelioid cells, and polymorphonuclear leukocytes are detected in cases with marked tubular injury that display tubulitis. Another feature of tubular injury is the phenotypic change of their lining epithelial cells into migrating fibroblasts, so-called epithelial-mesenchymal transition (EMT) (
Fig. 35.1D), an event considered to herald the onset of fibrosis.
15 Even in the absence of evident epithelial cell injury on light microscopy, an electron microscopy reveals structural cell changes, the loss of the brush border, and the fragmentation or lamination of the TBM. As a rule, immunofluorescent studies are not revealing. Scant and nonspecific granular staining for immunoglobulins, usually without complement, may be detected along the TBM. In rare drug-induced cases, linear deposits of immunoglobulin G (IgG) and complement (C3) may be present, indicating antibodies directed against tubular membrane antigens.
1,
8,
9There is increasing evidence for a participatory role of the epithelial cells in the pathogenesis of ATIN.
8,
9,
13,
14,
15 The enhanced expression of the human leukocyte antigen (HLA) class II antigens (HLA-DR) of injured tubular epithelial cells has been demonstrated, but shows no correlation with the intensity or phenotype of the infiltrating interstitial cells.
20 The overall profile of immunocompetent cells identified in ATIN suggests an important pathogenetic role for cell-mediated immunity in which the epithelial, vascular, and infiltrating cells are active and participating components.
8,
9,
21,
22,
23,
24
PATHOGENESIS
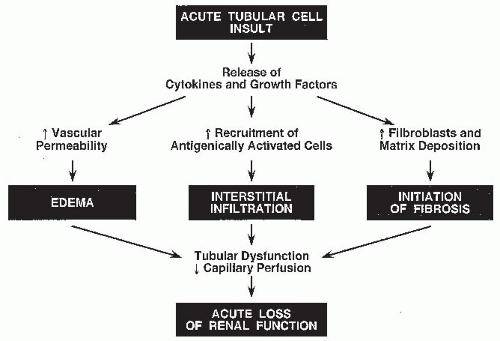
The immune response implicated in the pathogenesis of ATIN can be categorized into three phases: an antigen expression and recognition phase, an integrative or regulatory phase, and an effector or mediator phase (
Fig. 35.2). In the first phase, either the resident peritubular interstitial cells or the injured tubular epithelial cells, which express major histocompatibility complex (MHC) class II, may function as antigen presenters.
1,
25The subsequent integrative or regulatory phase may suppress or intensify the reaction to the stimulus provided by the antigen-presenting cells. This is a rather complicated phase of the immune response, in which T cells and antibodies directed at the presented antigen play a central role. Locally produced cytokines implicate a participatory role for the resident interstitial, tubular epithelial, and vascular pericytes and endothelial cells in this regulatory phase.
13,
14,
24,
25 The exposure of tubular epithelial cells (TECs) to selected insults or toxins results in the activation of the TECs and their expression of various growth factors and chemokines, such as interleukins (IL), monocyte chemoattractant proteins (MCP), MHC, vascular endothelial growth factor (VEGF), transforming growth factor (TGF), and regulated on activation, normal T expressed and secreted (RANTES).
26,
27 In this phase, the subsequent bidirectional cross-talk between the
TECs and the recruited infiltrating inflammatory cells, either by soluble factors or by direct contact, ultimately modulates the course of renal involvement and its potential for reversibility.
The final effector or mediator phase is mediated primarily by humoral factors released by the infiltrating cells, with the TECs and vascular pericytes playing a participatory role. The cytotoxic cells may induce injury by the release of proteases, and the inducer cells may do so by the release of lymphokines, which in addition to a direct detrimental action, augment the role of the macrophages. In turn, the release of collagenases, elastases, and reactive oxygen species by the macrophages magnifies the injury initiated by the lymphocytes. Lymphokines also promote fibroblast proliferation and alter the balance in favor of increased matrix synthesis rather than removal.
8,
13,
14,
26In the final analysis, it is the presence of a rather wide range of activated mononuclear cells and their interaction with each other and with renal parenchymal cells during the integrative and regulatory phases that is potentially damaging to the kidney (
Table 35.1). Several of the cellular (epithelial, endothelial, lymphocytes, macrophages) signals have varying, often overlapping, functions that interact to modulate or amplify the inflammatory reaction of ATIN. Some of those that have been identified include cell surface markers that have either
antigenic (MHC, HLA class II, secreted protein acidic and rich in protein [SPARC]) or
adhesive (intercellular [ICAM-1] and vascular [VCAM-1] adhesion molecules, integrins, selectins) properties. Others are
chemoattractant MCP-1; osteopontin; macrophage inflammatory protein-1 (MIP-1); eotaxin; RANTES,
proinflammatory (interleukin [IL]-6 and IL-8; TGF-β; platelet-derived growth factor-β [PDGF-β]; granulocyte-monocyte colony-stimulating factor [GM-CSF]; tumor necrosis factor-α [TNF-α]),
vasoactive (adenosine; nitrous oxide [NO]; endothelin-1 [ET-1]),
cytotoxic (metalloproteinases [MP]; tissue inhibitor metalloproteinases [TIMP-1]; reactive oxygen radicals; ferric ion), or
fibrogenic [TGF-β, PDGF, IL-1, IL-6, TNF, plasminogen activator inhibitor (PAI)]. Evidence also exists for possible protective cytokines that can favorably modify the proinflammatory sequence of events.
27 The resultant integration, during the regulatory and effector phases, either suppresses the effector phase, as in mild forms of ATIN, or amplifies it, as in severe forms of ATIN. Ultimately, feedback mechanisms and the removal of the inciting agent restore the response to injury to its baseline dormant state with consequent recovery of normal kidney function or residual permanent damage.
1,
18,
24,
28,
29,
30,
31The bulk of the available information about the phases of the immune response and the interstitial inflammatory reaction in ATIN derives from studies in experimental animals, particularly in models of recombinant rats and mice.
9,
26,
29,
32 Unfortunately, there are no animal models that correspond to the reaction that occurs in human ATIN. A principal limitation of human studies is that the data derived from kidney biopsies provide information only at fixed moments in the course of a lesion that is an evolving process. The same limitation applies to the serologic data provided in clinical studies that are obtained at a fixed period, usually when the lesion is well established and often at its worst, with limited or no data available on the onset or resolution phases of the lesion. These limitations notwithstanding, considerable evidence has accrued for a role of the principal immunologic mechanisms—cell-mediated injury, anti-TBM antibodies, immune complex deposition—in the pathogenesis of ATIN. Although there is evidence that all three mechanisms may be operative to varying degrees in different patients, the bulk of the available information favors a predominant role for cell-mediated injury.
8,
9,
25Contrary to the extensive experimental data in support of anti-TBM disease from animal models of interstitial disease, anti-TBM antibodies have been detected only rarely
in human ATIN.
1 The dearth of evidence for anti-TBM-mediated disease is far from convincing for an important role, if any, of this mechanism in human ATIN. The same limitations apply for a role of immune complex-mediated ATIN. The cases in which granular deposits of IgG have been detected have been in patients with Sjögren syndrome
32 and systemic lupus erythematosus,
29,
30 the underlying disease mechanism of which accounts for the deposition of immune complexes in the kidneys as well as other body organs. Granular deposits in other, more common forms of ATIN are rare and never a dominant feature.
By contrast, the evidence in favor of cell-mediated disease is overwhelming. The infiltrating cells are antigenically activated. T-cell reactivity has been demonstrated from in vitro studies of lymphocyte stimulation.
19,
22,
32,
33,
34 Comparative studies of ATIN due to NSAIDs or antibiotics have failed to reveal significant differences in the overall percentage composition of T cells, B cells, and monocytes.
20 The reported differences in infiltrating cell subtypes can be due to individual genetic background, the nature of the insult, and the point in time during the course of the disease when biopsies were obtained. Nevertheless, it is clear that where sought, activated antigens have been demonstrated on the surface of the infiltrating cells, and plasma cells containing IgE, IgA, IgG, and IgM have been detected. However, no diagnostic pattern of markers or infiltrating cells has emerged.
1,
20
FUNCTIONAL MANIFESTATIONS
The principal manifestations of ATIN are those of tubular dysfunction, which is one reason that the term tubulointerstitial nephritis was introduced in preference to the initial appellation of interstitial nephritis. As a rule, tubular dysfunction precedes the onset of azotemia, which in turn precedes that of oliguria. As such, vigilance to clinically evident abnormalities of tubular function, including polyuria, is essential to the early detection of ATIN during its initial reversible stages.
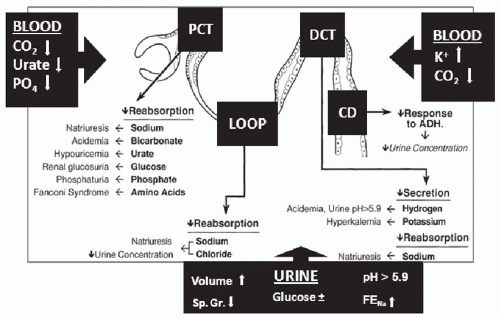
Because of the focal nature of the lesions and the segmental nature of normal tubular function, the pattern of tubular dysfunction that results varies depending on the major site of injury, whereas the extent of damage determines the severity of tubular dysfunction (
Fig. 35.3). The principal hallmarks of glomerular disease, such as salt retention, edema, and hypertension, characteristically are absent. However, massive proteinuria does occur in ATIN as a result of NSAIDs use and, occasionally, antibiotic use.
8,
9,
10,
34 In addition to the direct injury to epithelial cells that may account for tubular dysfunction, changes in the interstitial volume and composition contribute to the functional abnormalities that develop. A major part of the reabsorbed or secreted tubular fluid has to traverse a true interstitial space.
35 The structure, composition, and permeability characteristics of the interstitial space must, out of necessity, exert an effect on such an exchange. A change in any of these parameters of the interstitium or those of the epithelial cell tight junctions, by delaying equilibration or exchange processes, could account for the functional alterations of the renal tubule that develop in ATIN (
Fig. 35.3). It also is possible that changes in the pressure of the supporting interstitium, which are transmitted to the periarterial space, could affect the blood
flow to the adjacent tubule and thereby cause further tubular dysfunction. Tubular alterations also may be reflected in reductions of the GFR through tubuloglomerular feedback as the function of the affected proximal tubular segments becomes compromised and both the rate and the load of solute delivery to the macula densa are altered.
1 Furthermore, the increase in hydrostatic pressure of the edematous interstitium, as has been demonstrated in allograft rejections, may adversely affect the intra-arteriolar pressure and may cause a reduction of the hydrostatic pressure transmitted to the glomerular capillaries. In addition, a vasoactive effect of the cytokines, eicosanoids, and reactive oxygen species elaborated by the interstitial infiltrating cells and injured epithelial cells may affect renal hemodynamics through the increased production of vasoconstrictive substances, such as thromboxane A
2 and leukotrienes, and through the decreased production of vasodilators, such as nitric oxide, by the injured proximal tubule cells.
36,
37,
38 Evidence for a role for cytokines released by the infiltrating cells in the reduction of renal blood flow and GFR has been advanced from studies of an experimental model of acute obstructive nephropathy that is characterized by acute mononuclear cell infiltrates.
39The tubulointerstitial lesions usually are principally localized in the cortex and corticomedullary junction. Cortical lesions predominantly affect either the proximal tubule or the distal tubule. Corticomedullary lesions affect the loop of Henle and the medullary collecting duct. The change in the normal function of each of the affected segments then determines the manifestations of tubular dysfunction (
Table 35.2 and
Fig 35.3). Lesions principally affecting the proximal tubule result in bicarbonaturia (proximal renal tubular acidosis), glucosuria (renal glucosuria), aminoaciduria, β
2-microglobinuria, phosphaturia, and uricosuria.
1 The latter two events can be valuable in suggesting the possibility of tubulointerstitial disease when the serum phosphate and urate levels are lower than expected in any patient with reduced kidney function. The presence of glucosuria when blood sugar levels are normal should always lead to a consideration of ATIN. The distal nephron segment secretes hydrogen and potassium and regulates the final amount of sodium chloride excreted. Lesions primarily affecting the distal tubule will result in the distal form of renal tubular acidosis, hyperkalemia, and salt wasting. Lesions that primarily involve the corticomedullary junction disproportionately affect the loops of Henle, the collecting ducts, and other medullary structures essential to achieving and maintaining medullary hypertonicity, resulting in different degrees of nephrogenic diabetes insipidus, polyuria, and nocturia. Because of the reduced proximal tubular reabsorption and the disrupted medullary function, a polyuric phase almost always precedes the onset of oliguria in ATIN.
1Although this general framework is useful in localizing the site of injury, considerable overlap is encountered
clinically with different degrees of proximal, distal, and medullary dysfunction present in any one patient, all of which usually precede any clinically detectable changes in GFR. In most cases, however, the reduction in GFR and consequent azotemia are the presenting clinical abnormalities calling attention to the acute kidney injury. Preceding or coexistent evidence of tubular dysfunction, unless specifically sought, may go undetected and may either delay the diagnosis or result in the wrong clinical diagnosis. One can only speculate on the number of cases of ATIN that go undetected clinically because of the presence of tubular dysfunction alone in the absence of significant azotemia or oliguria—hence the importance of monitoring for tubular dysfunction in patients exposed to agents known to be associated with ATIN, and the necessity of documenting tubular dysfunction in those with mild azotemia to marshal evidence for the diagnosis of ATIN, before its progression to kidney failure (
Table 35.2). Importantly, it is at this early stage of the disease that the lesions of ATIN are potentially reversible and responsive to therapy.
8,
9,
40
CLINICAL FEATURES
The clinical presentation of individual cases of ATIN is diverse and varies to some degree depending on the causative factor (
Table 35.3). The manifestations best characterized in methicillin-induced ATIN can be considered the prototypically classic clinical manifestations of a hypersensitivity reaction, around which general variation occurs depending on the causative agent and individual variation occurs depending on the particular case encountered clinically.
8,
9,
21,
25In most cases, symptoms develop several days or even weeks after exposure to the inciting agent, which in the case of drugs, is not dose dependent. The classic triad of low-grade fever (35% to 70%), fleeting skin rash (25% to 40%), and mild eosinophilia (35% to 60%) described with methicillin-induced ATIN is not always present, and certainly it is less common for all three to occur together. Their documentation depends to some degree on the vigilance with which they are sought because they may be mild and transient. The full triad was noted to be present in approximately 20% of cases of methicillin-induced ATIN, but the triad was noted only in less than 10% of cases of ATIN induced by other drugs in which any one of them (fever, rash, or eosinophilia) is less likely to occur than with methicillin.
8,
9,
11,
41 The skin rash consists of erythematous maculopapular lesions, which often are pruritic, and preferentially affect the trunk and the proximal portion of the extremities. Flank pain or a sense of fullness, reflecting edema-induced distention of the renal capsule, may be present in over one-third of the cases when queried and may be the presenting symptom in some cases.
42 Gross hematuria, another presenting feature, may be present in 5% to 15% of drug-induced ATIN cases. A transient
eosinophilia, often in methillin-induced ATIN (˜60%) and less commonly with other drugs (˜30%), is present, but may go undetected unless specifically sought. A history of arthralgia may be elicited in more than one-fourth of cases.
8,
9,
21A urinalysis can be helpful in the diagnosis (
Table 35.3). Proteinuria, hematuria, and pyuria are present in most cases. The proteinuria is mild, seldom exceeds 2 g per day, and only rarely is in the nephrotic range, except in those cases due to NSAIDs.
8,
9,
11 Microscopic hematuria is present in 70% to 90% of cases; rarely, red blood cell casts may be detected. The pyuria is nonspecific except when eosinophils are detected in appropriately prepared and carefully examined urinary sediment.
1,
8,
9 White blood cell casts occur and are fairly characteristic when they contain eosinophils.
43 Although eosinophils may be observed with a Wright stain of well spun urinary sediment, the use of a Hansel stain on the sediment of an alkalinized urine sample is superior in detecting eosinophils. The mere detection of eosinophiluria is not specific for ATIN.
1,
44 The sensitivity of eosinophiluria for the diagnosis of ATIN has been estimated to be about 60% and its specificity has been estimated to be 85%, with a positive predictive value of 38%. These are figures derived from retrospective chart reviews of cases of clinically suspected ATIN but which were not established by a kidney biopsy or definite etiology.
44,
45 Eosinophiluria is present in approximately 15% of hospitalized patients, in whom it usually is caused by a variety of other inflammatory diseases of the urinary tract, and in only 14% of those with eosinophiluria is it due to ATIN.
44,
45 The presence of eosinophiluria is a better predictor of ATIN when more than 5% of the urinary leukocytes are eosinophils, in which case 40% of the eosinophiluric patients have been estimated to have ATIN, as opposed to an incidence of 3.5% in those in whom less than 1% are eosinophils.
44,
45 Other conditions in which an eosinophiluria exceeding 5% may be present are urinary tract obstruction, cystitis, contrast-induced AKI, IgA nephropathy, and cholesterol emboli. Thus, the detection of eosinophiluria, although useful, is neither necessary nor sufficient for the diagnosis of ATIN. It is regrettable that compared to urine examinations for eosinophils in the diagnosis of ATIN, it is rare that attempts are made for the detection of specific tubular dysfunctions characteristic of ATIN such as renal glucosuria, increased fractional excretion of uric acid, an inability to concentrate the urine maximally, and to conserve sodium.
1The impairment in kidney function varies, ranging from discrete selective abnormalities of tubular function to frank kidney failure, with or without oliguria.
1,
8,
9,
11 As a rule, increments in blood urea nitrogen and serum creatinine develop after tubular dysfunction is detectable and while the patient is still nonoliguric or even polyuric. Oliguria develops if early features of ATIN, modest azotemia, decrements in estimated GFR, and evidence of tubular dysfunction go undetected and exposure to the offending agent continues. Kidney injury is more likely to occur in older patients and is more severe in those who become oliguric. The duration of the oliguric period is variable, ranging from a few days to several weeks. Supportive renal replacement therapy may be required in approximately one-third of those patients. Reversal of kidney injury and a return to baseline kidney function is the rule in the majority of cases (about 60% to 65%). Irreversible kidney injury can occur but is rare (about 5% to 10%), whereas partial recovery with a persistent impairment of kidney function is relatively more common (10% to 20%), especially in cases where interstitial fibrosis and granulomas are present in biopsy specimens.
8,
9,
11,
21Increased kidney size on ultrasonography, reflecting an interstitial edema, is common but nondiagnostic. Radioactive gallium uptake by the kidney, reflecting interstitial cellular infiltration, can be detected in one-third of cases, but lacks diagnostic specificity.
46 This modality may be particularly useful when a kidney biopsy is contraindicated or is difficult to perform in severely ill patients.
46 The lymphocyte stimulation
test can be valuable in the diagnosis of drug-induced ATIN, especially in determining the responsible agent in cases of multiple-drug therapy.
29,
30 The serum level of IgE can be elevated and IgE-containing plasma cells have been demonstrated among the renal interstitial cellular infiltrates, providing further evidence for a hypersensitivity reaction.
25
INCIDENCE AND DIAGNOSIS
The frequency with which ATIN accounts for cases of clinically encountered AKI is difficult to establish. The diagnosis of ATIN is based on the finding of characteristic morphologic features on a kidney biopsy and on the identification of the causative factor. Both of these requirements for a correct diagnosis are fraught with difficulties and limitations, leading to the overall underdiagnosis of ATIN.
Part of the difficulty associated with diagnosing ATIN stems from the relatively low index of suspicion with which its possibility is considered clinically and the general reluctance to perform a kidney biopsy in cases of AKI in general, and in cases of ATIN in particular when symptoms subside after the discontinuation of the causative medications.
7 Almost all cases of ATIN that have been biopsied have been in renal failure. Even if a biopsy is performed, there are difficulties in differentiating the lesions of ATIN from those of ATN, which is a distinct possibility in most cases of AKI in which the prevailing clinical condition could be conducive to either ATIN or ATN.
1,
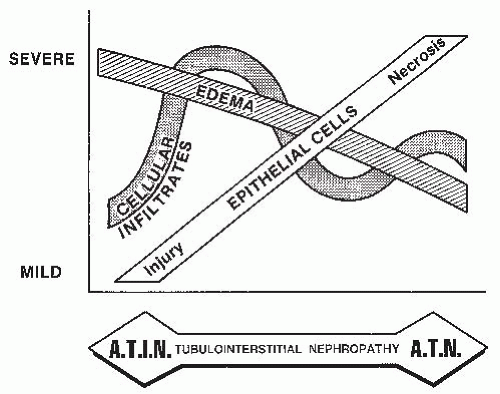
6 An essential differential feature between the two entities is the magnitude of interstitial edema and cellular infiltrates, which are more prominent in ATIN, and the magnitude of tubular cell injury, which is more prominent in ATN (
Fig. 35.4). However, given the variable degree to which each of these features may be present in each entity, there is sufficient overlap between the extent of edema and the severity of tubular injury in ATIN and ATN to make it difficult to differentiate among them on morphologic features alone, at least in some cases.
6,
45 There also are difficulties associated with identifying the causative factor. Clinically, the removal of a suspected agent followed by the reversal of the lesion strongly suggests the diagnosis of ATIN. This can be particularly convincing in the presence of systemic manifestations of a hypersensitivity reaction, such as fever, skin rash, and eosinophilia in addition to AKI, all of which subside on the removal of the inciting factor or agent. Difficulties arise when the only manifestation is AKI and when a number of corrective measures are instituted simultaneously. Moreover, most patients are on several drugs, and there is the expected tendency to incriminate the most common and better known agents that have been associated with ATIN.
8 The diagnostic limitations of removing agents wrongly surmised as a cause of ATIN have been shown by studies in which drug-induced lymphocyte stimulation testing (DLST) revealed causative agents that had not been suspected clinically.
34Most of the figures quoted in the literature on the incidence of ATIN stem from retrospective studies based on kidney biopsies. A review of unselected kidney biopsy specimens reveals a low incidence of ATIN. A review of biopsy specimens from patients with unexplained AKI reveals a frequency that ranges from 8% to 22%,
7,
47,
48,
49,
50 which generally is higher in the elderly population.
51 Thus, given the very small number of patients with AKI who are subjected to a kidney biopsy, the necessity of a kidney biopsy to diagnose ATIN, and the failure to suspect the diagnosis of ATIN clinically all render conclusions derived from biopsy reports of limited value in estimating the true incidence of ATIN.
7 Furthermore, because of the variable and often subjective reasons for which kidney biopsies are done in different centers, any attempt to compare or combine results from different reports would not be useful. In cases of clinically evident AKI, ATIN probably accounts for approximately 15% to 20% of them.
7,
8,
9