Given the biochemical similarity between myoglobin and hemoglobin, the general consensus that they share a common pathogenetic pathway, and the fact that the classical animal models used to study pigment-induced oliguric AKI share intravascular hemolysis (hemoglobinuria) and rhabdomyolysis (myoglobinuria), the pathogenesis of these two pigments are presented together in this discussion.
Hypovolemia and Renal Ischemia
During the initial phase of glycerol-induced AKI (an animal model for rhabdomyolysis), there is a marked reduction in cardiac output (36%), renal blood flow (RBF) (20%), and an increase in renal vascular resistance.
30 Subcutaneous or intramuscular (but not intravenous) glycerol not only produces muscle injury but also causes sequestration of fluid into the injection site.
31 Thus, the hemodynamic changes are due, in part, to the migration of plasma water into the site of injury with consequent severe intravascular volume contraction occurring in this model of myohemoglobinuric AKI. Comparable hemodynamic changes occur in clinical settings that damage and cause necrosis of the skeletal muscle such as crush syndrome.
7 Moreover, the conditions that predispose one to rhabdomyolysis, such as drug-induced coma with accompanying poor oral intake, or excessive insensible fluid losses from exhaustive exercise or burns, contribute to intravascular volume depletion and compromise of renal function.
In the initial phases of glycerol-induced AKI, the reduction in RBF is associated with a redistribution of regional blood flow from the outer to the inner cortex
32 and vasoconstriction of the afferent and efferent arteriole. The proposed mediators of this initial renal vasoconstriction include (1) increased sympathetic nerve activity, (2) augmented activity of the renin-angiotensin system, (3) reduced nitric oxide production and availability, (4) suppressed renal prostaglandin production, (5) increased plasma vasopressin concentration, and (6) glomerular microthrombi.
33 The reduction in nitric oxide may be due to the fact that heme proteins can scavenge this important endogenous vasodilator. Nitric oxide synthase inhibition worsens and nitric oxide supplementation protects against glycerol-induced AKI. This lends support to the protective effects of nitric oxide in the pathogenesis of myoglobin-induced AKI.
34The critical role that intravascular volume depletion plays in the pathogenesis of myohemoglobinuric AKI is demonstrated by studies in which volume status is manipulated in the glycerol-treated rat. In initial studies, Oken et al.
35,
36 noted that renal damage was ameliorated if the rats ingested adequate quantities of water before the administration of glycerol. Similarly, Hsu et al.
30 found that the reduction in RBF and function in response to the administration of glycerol was attenuated in rats chronically drinking saline compared with rats drinking water. Reineck et al.
37 provided a better understanding of the important temporal relationship
between volume expansion and improvement of renal function in the glycerol-treated rat. Like other investigators, they noted a significant reduction in RBF and GFR after the administration of glycerol. These variables could be restored to normal levels by volume expansion with Ringer’s solution at 3 and 6 hours, but not at 18 hours after the administration of glycerol. They concluded that the initial decrease in GFR and the low fractional excretion of sodium was due to a decrease in RBF (renal hypoperfusion), whereas other events (e.g., tubular necrosis) accounted for decreased GFR at later time points. These preclinical studies support the clinical observation that, initially, patients with myoglobinuric AKI have features of prerenal azotemia, including a low urinary fractional excretion of sodium.
38 In addition, they provide the rationale for early use of volume expansion in patients with rhabdomyolysis and hemoglobinuria.
Myoglobin and Hemoglobin Nephrotoxicity. Bywaters and coworkers
2,
3 expanded on their original description of the clinical syndrome of rhabdomyolysis-induced AKI to examine the role of myoglobin as a direct nephrotoxin. They noted that rabbits ingesting an acid diet with a urine pH below 6.0 had AKI after the infusion of human myoglobin, whereas rabbits ingesting a normal diet were spared from renal injury.
3 Other investigators
39,
40 have confirmed this observation that intravenous infusions of myoglobin are relatively benign but can become highly nephrotoxic in the setting of acidemia/aciduria and volume depletion. Vetterlein et al.
41 demonstrated that infusions of myoglobin had no effect on RBF in normal rats, but worsened RBF in hypotensive animals. Thus, it appears that heme proteins can intensify the degree of vasoconstriction in the setting of hypovolemia. This may explain the clinical observation that the mere presence of myoglobinuria or a markedly elevated creatine kinase at the time of hospital admission had little predictive value in determining who experiences AKI.
17 These observations suggest that other conditions (i.e., volume depletion, acidemia) are required for renal injury to occur.
To address the question of why heme pigments are nephrotoxic only in certain metabolic conditions, Braun et al.
31 investigated the effect of breakdown products of heme pigments on renal tubular transport. First, they noted that 4 hours after a subcutaneous glycerol administration to rats there was both swelling and pallor of the proximal tubule and depression of normal tubular uptake of hippurate and tetraethylammonium. The investigators measured the uptake of hippurate in renal cortical slices incubated with various specific heme proteins or their derivatives and found that incubation with hemoglobin did not depress uptake if the pH of the medium was kept at 7.4. However, uptake was depressed when the pH was lowered to 5.4 or during hypoxic conditions. In an acidic medium (pH <5.6), both myoglobin and hemoglobin dissociate into ferrihemate (hematin; molecular weight, 670 Da) and their respective globin moieties.
42 Incubation with ferrihemate, regardless of the pH of the medium, depressed the uptake of hippurate in the renal cortical slices, whereas incubation with either globin or albumin alone had no significant effect on transport. The inhibitory action of ferrihemate on hippurate transport could be mitigated if the incubation medium also contained albumin, which presumably bound the ferrihemate. Intravenous injection of ferrihemate has been shown to cause glomerular and tubular damage in the dog.
43 Therefore, it has been proposed that after filtration by the glomerulus, myoglobin or hemoglobin is converted to ferrihemate in the presence of an acid tubular fluid, or after exposure to the acid pH of cellular lysosomes, and it is this metabolite that is directly nephrotoxic.
These and other studies implicate the heme moiety as a potent pro-oxidant molecule.
44,
45 It is well established that free heme can facilitate the production of reactive oxygen species via Fenton/Haber-Weiss reactions. Under physiologic conditions, free heme is sequestered by heme binding proteins, and oxidative stress can cause the release of heme, thereby increasing free heme levels. In addition, evidence suggests that the iron component of heme is the culprit of heme-induced oxidative damage.
44,
45 The central role of iron has been substantiated by a number of studies demonstrating amelioration of both myoglobinuric and hemoglobinuric AKI and lipid peroxidation by the iron chelator, deferoxamine.
46 On the other hand, Zager
47 has also shown that deferoxamine attenuates renal damage in the glycerol-induced model of AKI, but concluded that iron toxicity is mediated by factors other than free radical generation. For example, it has been suggested that heme protein endocytosis in the proximal tubule sensitizes the tubular cell membranes to the damaging effects of phospholipase A
2.
48 In addition, heme proteins appear to deplete cellular ATP stores and, thus, have an adverse effect on cellular energetics.
5 Iron toxicity may be due to redox cycling of the heme moiety from ferrous to ferric and to ferryl oxidation states.
49In order to contend with the pro-oxidant heme moiety, the kidney induces antioxidant defensive machinery, including heme oxygenase-1 (HO-1).
44,
45 HO-1 catalyzes the ratelimiting step in the oxidative degradation of heme liberating equimolar amounts of iron, carbon monoxide, and biliverdin. Iron in turn induces the expression of ferritin. HO-1 is known to have important antioxidant, anti-inflammatory, and antiapoptotic functions that have been attributed to one or more of its byproducts.
44,
45 Nath et al.
50 have demonstrated that the renal induction of both HO-1 and ferritin is increased in the glycerol-induced model of myohemoglobinuric AKI. Prior induction of HO-1 coupled with increased ferritin synthesis attenuated renal damage, whereas pharmacologic inhibition of the enzyme or its gene deletion worsened renal function.
50,
51 This increased activity of HO-1, or possibly a broad-based proximal tubular cytoresistance in the kidney, may explain the experimental observation that after induction of myohemoglobinuric AKI rechallenging the animals with a second dose of glycerol does not result in AKI.
52 One speculation is that in the setting of clinical myoglobin-induced AKI, there may be factors contributing to the inhibition of HO-1 and ferritin synthesis, or a
diminution in proximal tubular resistance, resulting in both an accumulation of nephrotoxic iron and in tubular necrosis.
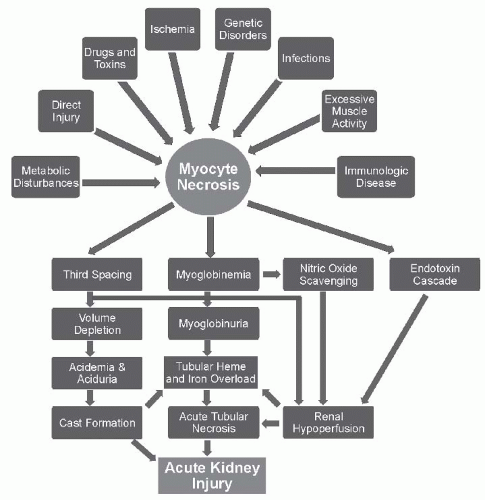
Tubular Obstruction. Filling of the tubular lumen by pigmented casts that become inspissated and obstruct urinary flow with subsequent injury to tubular epithelium is one of the earliest mechanisms proposed to explain the nephrotoxicity of the heme pigments.
53 In their original clinical description of rhabdomyolysis-induced AKI, Bywaters and Beall
2 described the prominent histologic features, including the appearance of tubular obstruction by cellular debris and pigmented casts. It has been suggested that hypovolemia and academia, and the concomitant acidic concentrated urine, facilitate the precipitation of filtered myoglobin or hemoglobin leading to obstructive cast formation.
54 The presence of the Tamm-Horsfall protein in the tubular lumen is critical for heme protein cast formation in the distal nephron. Moreover, an obstructing cast induces urinary stasis, providing for an extended time for proximal tubular heme reabsorption and its attendant tubular toxicity, as noted previously.
55
Tubular obstruction can decrease GFR either by increasing the tubular pressure and thus decreasing the glomerular transcapillary hydraulic pressure, or by inducing the release of factors (e.g., thromboxane) that cause renal vasoconstriction, thereby reducing glomerular blood flow. The importance of tubular obstruction as a possible mechanism of heme pigment-induced AKI is suggested by the studies of Zager
47 that explored the reasons why mannitol exerts a protective effect against this syndrome. The major beneficial effect of mannitol was attributed to its diuretic effect, which presumably decreased cast formation and proximal tubular uptake of heme proteins. Similarly, alkalinization of the urine may mitigate against myoglobinuric AKI by increasing the solubility of myoglobin (reduced cast formation) and inducing a solute diuresis.
54Although there is evidence that tubular obstruction may be a factor in the pathogenesis of the AKI, it probably is not the primary cause of the initial decrease in GFR in myohemoglobinuric AKI. Rather than high intratubular pressures from obstructing casts, intratubular pressures were found to be low in the glycerol-induced model of AKI.
35 This observation was interpreted to indicate that the presence of casts is the result, rather than the cause, of the decrease in GFR and urine flow. Instead of causing the initial decrease in renal function, cast formation may play a role in the maintenance of the renal failure once it develops.
56Glomerular Fibrin Deposition. Because of the liberation of tissue factors, both rhabdomyolysis and intravascular hemolysis can initiate disseminated intravascular coagulation (DIC).
19 Fibrin strands have been demonstrated in glomeruli from patients
57 and experimental animals
58 with rhabdomyolysis-induced AKI. Intravenous infusion of a muscle extract in rabbits resulted in DIC, renal dysfunction, and glomerular microthrombi, whereas an intravenous infusion of pure myoglobin had no untoward effect.
59 This led to the conclusion that myoglobin, per se, is not the primary cause of the coagulation cascade activation in the crush syndrome, but rather it is the release of other muscle constituents that induces DIC and the subsequent deposition of glomerular microthrombi that are responsible for rhabdomyolysis-induced AKI.
Clinical and Laboratory Features of Rhabdomyolysis and AKI
The diagnosis of myoglobinuria can be suspected from a history and physical examination. However, the clinical features of rhabdomyolysis are nonspecific and the course of the syndrome is quite variable depending on the underlying cause and the general condition of the patient. The syndrome has local as well as systemic features and early or late complications may occur. Because the prompt recognition of rhabdomyolysis is critical to preventing late complications, all suspected cases must undergo a complete clinical inquiry, observation, and laboratory follow-up.
Risk Factors for Acute Kidney Injury. The frequency of AKI in the setting of rhabdomyolysis is unknown, and reports of frequency have ranged from 13% to 50%.
1 Gabow and colleagues
17 emphasized that no single laboratory value could predict which patients are at high risk for the development of AKI. However, using discriminant analysis, patients could be separated into high- and low-risk groups, with the high-risk group (elevated serum potassium and creatinine and reduced serum albumin concentrations) having a 41% prevalence of AKI.
Based on a large historical cohort (157 patients), Ward
60 identified clinical and laboratory differences between those patients in whom renal failure did or did not develop, and factors predictive of progression to renal failure. As shown in
Table 36.2, patients with rhabdomyolysis and renal failure were older, had a higher incidence of hypertension, and were more hypotensive and volume depleted. A significantly greater proportion of them had a creatine kinase level greater than 16,000 IU per liter, although elevations to this degree were seen in 10.7% of patients in whom renal failure did not develop (
Table 36.3). The renal failure group also had significantly higher serum potassium and phosphorus levels and lower serum calcium and albumin concentrations, and was more acidemic with a concomitant lower urinary pH. Sepsis, burns, and drug ingestion were the causes of rhabdomyolysis more closely associated with the development of renal failure. Using multiple logistic regression analysis, a scoring system was developed predicting the risk of renal failure in patients with rhabdomyolysis based on the variables of serum phosphorus, potassium, albumin, and creatine kinase concentrations, and the presence of volume depletion and sepsis. A point score of 7 or higher predicted a greater than 50% likelihood for the development of renal failure. In a multivariate analysis of 72 consecutive patients with rhabdomyolysis due to illicit
drug use, patients with a creatine kinase level greater than 25,000 IU per liter, hypotension, and leukocytosis were at a greater risk of developing AKI, whereas hyperthermia (temperature >38.5°C) was associated with a reduced risk.
61 This association does not indicate that hyperthermia is protective against rhabdomyolysis, rather it is most likely due to earlier presentation to, or evaluation or fluid resuscitation in the emergency department.
Urinalysis. Examination of the urine provides the first laboratory clue to the presence of myoglobinuria. Classically, the initial urine is dark (
Table 36.4) and usually with an acid pH; the benzidine or orthotoluidine reagent gives a positive reaction for blood (3+ to 4 +), but microscopic examination of the urinary sediment fails to reveal any red blood cells (RBCs). Specific tests for urine myoglobin determination are available in some clinical laboratories but, as noted earlier, urine myoglobin levels are not the most sensitive clinical markers for rhabdomyolysis. Although the strongest clinical clue for myoglobinuria is the presence of strongly heme-positive urine and the absence of RBCs, in one major series
17 hematuria was present in 32% and the dipstick was heme negative in 18% of the patients with rhabdomyolysis. In addition, proteinuria was detected by dipstick in 45% of patients,
17 which may be attributed to altered glomerular permeability or tubular transport of small proteins.
62 The urinary sediment demonstrates brown “debris” and, with the evolution of renal injury, pigmented brown granular casts and renal tubular epithelial cells are seen.
Serum Potassium Concentration. The most life-threatening consequence of rhabdomyolysis is the release of large amounts of intracellular potassium into the circulation. Given the crucial role that potassium plays in maintaining the homeostasis of resting membrane potential, it is evident that vital organs such as the heart are at greatest risk to sustain arrhythmogenic activity. This implies that an electrocardiographic follow-up is mandatory to monitor for potentially grave arrhythmias. Because more than 98% of total body potassium resides in cells, and skeletal muscle represents 60% to 70% of the total cellular mass, breakdown of even a small area of skeletal muscle releases a considerable potassium load. The presence of
acidosis may shift more potassium extracellularly and worsen the hyperkalemia. As noted in the previous section on Risk Factors for AKI, admission serum potassium levels tend to be higher in patients who go on to experience AKI.
60 Approximately half of an acute potassium load is handled by renal excretion
63; therefore, in AKI, serious hyperkalemia can result and is usually the major indication for dialysis.
Creatine Kinase. The classic laboratory finding of rhabdomyolysis is an elevated serum creatine kinase of at least five
times the normal value, where the striated muscle isoenzyme (CK-MM) is predominately found. The serum half-life of creatine kinase (˜36 hours) is much longer than myoglobin, which makes it a more reliable tool for diagnosis. Normal creatine kinase levels are 45 to 260 IU per liter. Following muscle injury, the level rises within 12 hours, peaks in 1 to 3 days, and declines 3 to 5 days after the cessation of muscle injury.
29 Although no correlation has been established between the absolute level of the creatine kinase and the risk for development of AKI, creatine kinase levels are significantly higher in patients in whom renal failure develops.
19,
29 Following admission, changes in creatine kinase concentrations provide some insight into whether the rhabdomyolysis is worsening or resolving, and following levels is essential to observe for the “second wave” phenomenon (described later in this chapter).
Acid-Base Balance. The conditions that cause rhabdomyolysis involve tissue trauma or ischemia and predispose one to an augmented acid load. In a study by Ward,
60 patients with rhabdomyolysis who progressed to renal failure tended to be more acidemic. An elevated serum anion gap is usual in patients with rhabdomyolysis and due to the impaired renal excretion of intracellular organic acids released from damaged muscles, as well as a retention of inorganic anions such as phosphate.
64
Uric Acid. Due to the release of intracellular purines from damaged myocytes, hyperuricemia is expected in patients with rhabdomyolysis, especially when the muscle injury is due to strenuous exercise or exertion.
Blood Urea Nitrogen: Creatinine Ratio. Both AKI and the increased release of creatine from damaged myocytes increase the serum concentrations of blood urea nitrogen (BUN) and creatinine. However, the rise in creatinine is more pronounced and, in turn, alters the normal 10:1 ratio of BUN to creatinine to a ratio of 6:1 or less. Based on creatine:creatinine kinetics and their respective concentrations in skeletal muscle, Oh
65 challenged this conventional view. He pointed out that the patient population in which rhabdomyolysis develops tends to have a larger percentage of younger men with a greater muscle mass, whereas other forms of AKI are more often associated with older and more cachectic patients who have less muscle mass and thus reduced creatinine production rates.
Calcium-Phosphorus Metabolism. The perturbations of calcium and phosphorus metabolism usually seen in most types of AKI appear to be exaggerated in rhabdomyolysisinduced AKI.
19,
66 Following the destruction of muscle cells, the release of inorganic phosphorus into the plasma causes hyperphosphatemia
19,
64 and subsequent hypocalcemia through the deposition of calcium phosphate in the destroyed muscle cells (dystrophic calcification) and other tissues. Hypocalcemia may be accentuated by the inhibition of renal vitamin D 1α-hydroxylase, which results in the downregulation of the production of the active form of vitamin D (1,25[OH]
2D
3). This observation may be explained by hyperphosphatemia, which is known to decrease synthesis of 1,25(OH)
2D
3 and to stimulate the production of the parathyroid hormone.
64,
67 A recent case report described elevated FGF23 levels in rhabdomyolysis-induced AKI and may provide a mechanism for the inhibition of renal 1α-hydroxylase.
68 Regardless of the mechanism, in the absence of frank tetany, hypocalcemia usually does not require treatment. In fact, correction of the hypocalcemia with vigorous intravenous calcium replacement may increase both dystrophic (calcium deposition in damaged muscle) and metastatic calcification due to the high serum PO
4 and the Ca × PO
4 product.
Approximately 20% to 30% of patients with myoglobinuric AKI experience transient hypercalcemia during the recovery (diuretic) phase.
19,
64 Early studies
69,
70 suggested that hypercalcemia was due to the normal remobilization of calcium deposits in the injured muscle that occurs during the recovery phase of AKI. Alternatively, it has been proposed that as renal function improves, the combination of a decreasing serum phosphorus concentration and the ambient secondary hyperparathyroidism, secondary to hypocalcemia, stimulates the synthesis of 1,25(OH)
2D
3 resulting in an “overshoot” hypercalcemia.
64 This augmented 1,25(OH)
2D
3 production may be due, in part, to the release of vitamin D from damaged muscle tissue.
64,
71Urinary Sodium Excretion. Impaired renal tubular reabsorption of sodium is typically seen in most types of oliguric AKI as manifested by a high fractional excretion of sodium. However, in both myoglobinuric and hemoglobinuric AKI, a low fractional excretion of sodium (<1%) has been observed
38 that resembles a prerenal azotemia during the early course. As noted earlier in this chapter, this phenomenon is most likely due to hypovolemia and vasoconstriction, which lead to renal hypoperfusion.
Disseminated Intravascular Coagulation. DIC is commonly present in patients with rhabdomyolysis and may be due to the release of intracellular thromboplastins that activate the clotting cascade.
63,
64 Moreover, DIC may be an important factor in the pathogenesis of the AKI (see section on Glomerular Fibrin Deposition, previously).