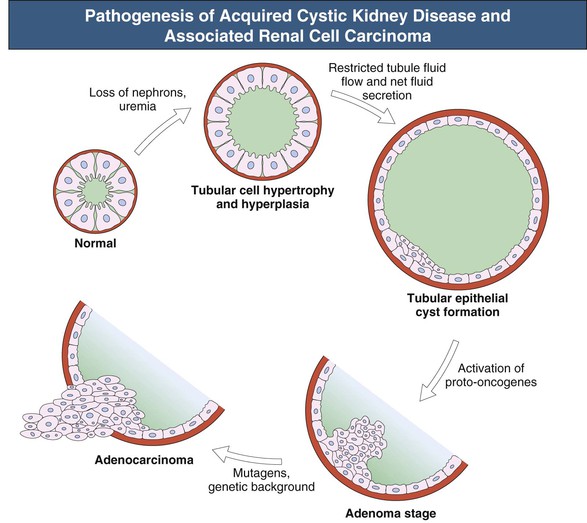
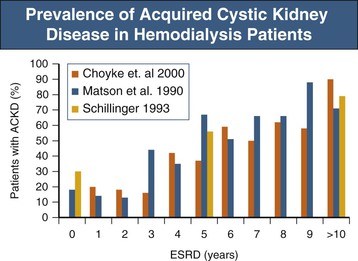
Anja S. Mühlfeld, Frank Eitner Acquired cystic kidney disease (ACKD) was first recognized in 1847 by John Simon in patients with chronic Bright disease. He described the development of cystic renal changes with cysts ranging from “mustard seed to as large as cocoa nuts” and also noted that they “run a slow and insidious progress during life, and often leave in the dead body no such obvious traces as would strike the superficial observer.” ACKD was “rediscovered” by Dunnill and colleagues1 in 1977 in kidneys from dialysis patients. Acquired cystic kidney disease consists of chronic kidney disease of any cause and must be differentiated from other types of cystic kidney disease (see Chapters 46 and 47). It is usually defined as more than three to five macroscopic cysts in each kidney of a patient who does not have a hereditary cause of cystic disease. ACKD is associated with renal neoplasms with such high frequency that some authors consider ACKD preneoplastic.2 Most cysts are lined by a single layer of epithelium composed of flat nondescript cells, cells with abundant cytoplasm and hyaline droplets, or small cuboidal cells resembling those from distal tubules or collecting ducts.2 Some authors have argued that the presence of a brush border on the luminal membrane suggests that the cysts arise primarily from proliferation of proximal tubular epithelial cells. Although the mechanisms of tubule transformation into cysts are not entirely clear, tubular epithelial cell hyperplasia is currently viewed as a central early event in ACKD pathogenesis (Fig. 89-1).3 Various factors have been implicated in the development of tubular hyperplasia, including plasticizer, ischemia, and uremic metabolites. However, the most important factor appears to be slow, progressive parenchymal loss, which could explain why the development or progression of ACKD does not appear to be influenced by the type of underlying renal disease or the choice of dialysis modality. The loss of intact nephrons is a strong stimulus for compensatory growth of the remaining, still intact nephrons, which is achieved by initial hypertrophy and later by hyperplasia. In these hyperplastic tubules, a cyst will develop if transepithelial fluid secretion continues, and if it is a result of anatomic distortion or obstruction, the distal outflow is impaired. With the continuing presence of mitogenic stimuli, the epithelial layer of the cyst becomes multilayered and atypical cells form intracystic papillary structures or mural adenomas. Activation of proto-oncogenes, chromosomal abnormalities, and additional factors such as genetic background, environmental chemicals, and sex hormones thereafter probably account for the transition of the proliferative process into malignant growth (see Fig. 89-1).3 Among patients starting maintenance dialysis treatment, prevalences of ACKD ranging from 5% to 20% have been described. ACKD can occur in some patients even before dialysis is initiated. In both chronic hemodialysis and peritoneal dialysis patients, prevalence then increases at a similar rate and reaches 80% to 100% after 10 years of treatment (Fig. 89-2).4–7 Children are also prone to the development of ACKD. Several but not all studies have reported an increased frequency or faster progression in men than in women. The rate of progression appears to slow after 10 to 15 years of dialysis. The frequency of ACKD as well as of renal tumors in dialysis patients may be underestimated on the basis of imaging methods alone. Renal cysts are detectable by ultrasound with a minimum size of 0.5 cm. Data obtained from 260 native nephrectomy specimens at the time of transplantation with a median dialysis duration of 1.0 year identified ACKD in 33%, renal adenomas in 14%, and renal cell carcinomas (RCCs) in 4% of the specimens.8 Acquired cystic kidney disease can manifest as unilateral or bilateral cysts, which are mostly cortical and variable in size and number. Rarely, severe ACKD can become macroscopically indistinguishable from adult polycystic kidney disease (PKD). In contrast to hereditary cystic diseases, the cysts of ACKD are strictly confined to the kidneys. The disease is usually asymptomatic and discovered accidentally during abdominal imaging procedures. Alternatively, it may manifest with potential complications or consequences of ACKD: ▪ Cyst infection, abscess formation, or sepsis. ▪ Erythrocytosis in advanced cases, similar to the erythrocytosis observed in PKD. Malignant transformation, the most feared complication of ACKD, accounts for about 80% of the renal cell neoplasms observed in uremic patients. In an unselected series of chronic dialysis or transplant patients, the cumulative incidence of RCC complicating ACKD is probably below 1%, although rates up to 7% have been reported in some small studies. These data indicate an up to 40-fold increased risk for RCC in ACKD patients compared with RCC in the general population. Risk factors include male gender (male-female ratio, 7 : 1), African American ethnicity, long duration of dialysis, and severe ACKD with marked organ enlargement. It is unknown whether the risk of malignant transformation differs between hemodialysis and peritoneal dialysis patients. About 85% of ACKD-associated RCCs are asymptomatic at the time of diagnosis. The remaining cases mostly manifest with bleeding, usually gross hematuria, from the tumor. In cases in which nephrectomy had to be performed in dialysis patients for intractable hematuria, RCCs not visualized before surgery were diagnosed in about one third of the patients. Compared with sporadic RCCs, ACKD-associated RCCs are characterized by younger age of the patient, male predominance, more frequent multicentric and bilateral manifestation, and less frequent metastases.2 Cystic changes in ACKD are typically bilateral but may vary between kidneys. Most ACKD kidneys are smaller than normal. An increase in size above normal can be observed in patients with cyst hematoma formation or malignant transformation. The cysts are usually restricted to the renal cortex. The size of the cysts ranges from microscopic to about 2 cm; about 60% of the cysts are smaller than 0.2 cm.2 Preneoplastic changes can be detected in ACKD kidneys, including atypical cyst-lining cells forming multiple cell layers and intracystic nodular formations. Up to 25% of kidneys with ACKD harbor tumors, about one third of which are carcinomas. RCCs arising from ACKD are multicentric in about 50% of cases and bilateral in about 10%. There is a unique histologic pattern in tumors arising in kidneys with ACKD, termed ACKD-associated carcinoma. They are characterized by a typical microcystic architecture, eosinophilic cytoplasm with Fuhrman grade 3 nuclei, and a frequent association with intratumoral oxalate crystals. In addition, these tumors commonly exhibit papillary architecture and clear cytoplasm.10 In patients with end stage renal disease without ACKD, the most common histologic type is clear cell carcinoma (44%) and the second most common is ACKD-associated carcinoma (23%).9 Clear cell carcinoma seems to be the most frequent type in patients with a dialysis duration of less than 10 years, whereas ACKD-associated carcinoma is the predominant tumor type in patients with dialysis duration of more than 10 years.11,12 Renal cell carcinomas in renal transplant patients tend to be smaller and to exhibit a lower T stage and a lower grade at diagnosis compared with patients with end-stage renal disease. Tumors are more often multifocal and bilateral, and the papillary subtype occurs more often.13,14 Because of the earlier stage at detection, the prognosis of RCC after renal transplantation generally is more favorable.
Acquired Cystic Kidney Disease and Malignant Neoplasms
Definition
Pathogenesis
Epidemiology
Clinical Manifestations
Acquired Cystic Kidney Disease–Associated Renal Cell Carcinoma
Pathology
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


