- Simple cysts are benign and do not cause symptoms. They do not need investigation or follow-up.
- Simple cysts can be multiple and not be part of a polycystic liver disease syndrome. The cysts do not cause symptoms and do not grow.
- Polycystic liver disease rarely causes liver failure. Symptoms are due to pressure, secondary infection, and very occasionally to the complications of portal hypertension. No treatment is required unless there are pressure symptoms.
Congenital Cysts in the Liver
A cyst is defined as a membrane-bound, fluid-filled structure. In autosomal dominant polycystic kidney disease (ADPKD) and polycystic liver disease (PLD), as well as in simple cysts, the epithelium lining the cyst is secretory, so that there is a tendency for cysts to enlarge over time. The cyst fluid has a composition similar to serum.
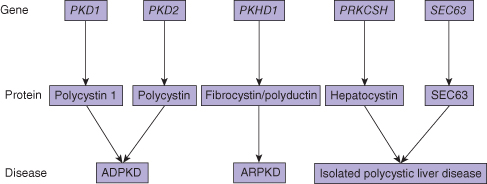
The inherited cystic diseases of the liver are all related to abnormal proteins in the cilia of the biliary epithelium (ciliopathy). The basic abnormality is defective cilial function, as a result of abnormal or absent membrane proteins. These are polycystin 1 and 2 (PC1 and PC2), hepatocystin, and fibrocystin/polyductin. PC1 is important in cell–cell adhesion. PC2 is probably a calcium channel protein. The precise functions of hepatocystin and fibrocystin/polyductin are unknown. The clinical phenotype varies, depending on the genetic abnormality. Similarly, whether the liver cysts are associated with kidney cysts also depends on the particular genetic abnormality (Fig. 25.1).
Fig. 25.1 Relationship between the genes, proteins, and diseases constituting congenital cystic conditions of the liver. ADPKD, autosomal dominant polycystic kidney disease; ARPKD, autosomal recessive polycystic kidney disease.
Congenital liver cysts occur as part of several syndromes. In order to understand the relationships between different cystic diseases in the liver it is necessary to discuss the embryology of the development of the biliary drainage system (Fig. 25.2). The bile ducts develop from the ductal plate, a cylindrical tube of cells surrounding a portal vein branch. The interlobular and intralobular bile ductules are formed by progressive remodeling after about week 12 of gestation. Arrest of maturation and lack of remodeling leads to persistence of an excess number of embryonic biliary structures (ductal plate malformation).1 This in turn results in the development of periportal fibrosis and dilatations in the bile ducts. Morphological studies demonstrate that the peripheral cysts of ADPKD and PLD arise from these biliary microhamartomas, but the centrally located cysts of Caroli disease arise from dilatations of the peribiliary glands in the liver.
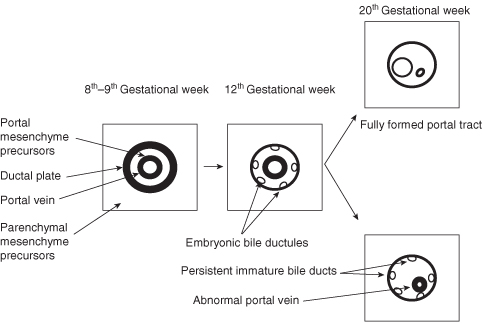
The liver cysts in ADPKD and PLD do not communicate with the biliary system. The cysts in Caroli disease do communicate with the biliary system.
Autosomal Dominant Polycystic Kidney Disease
Genetics
The prevalence of this entity is between 1 : 400 and 1 : 1000. The disease is characterized by progressive enlargement of cysts in the liver and kidney, leading to renal failure in about 50% of affected individuals. Most often the disease is caused by a mutation in the PKD1 gene coding for PC1, with about 20% being due to mutations in the PKD2 gene, coding for PC2. Liver cysts occur in about 60–75% of these patients. The frequency and severity of liver cystic disease is related to the severity of renal disease. Liver failure is rare, but can occur. The major hepatic manifestation is massive hepatomegaly as a result of large and enlarging cysts. The liver disease affects mostly females, and may have a relationship to female sex hormones.
Clinical
Polycystic liver disease causes symptoms because of the size of the liver. These may include early satiety, gastroesophageal reflux, mechanical back pain, and occasionally obstruction to the inferior vena cava, portal vein, or extrahepatic bile duct. Cysts may rupture, may bleed, or become infected but these are rare events.2
Diagnosis
The diagnosis of cystic liver diseases is usually made by ultrasonography. Computed tomography (CT) scanning provides similar information, but is usually easier for the non-radiologist to read.
Classification
Polycystic liver diseases are classified on the basis of number and size of the cysts and the amount of residual normal liver parenchyma between the cysts. Type I patients have a limited number (<10) of large cysts with large areas of non-cystic parenchyma. Type II PLD has diffuse involvement of liver parenchyma by medium-sized cysts with remaining large areas of non-cystic parenchyma. Type III patients have massive, diffuse involvement of liver by small and medium-sized liver cysts with little normal liver parenchyma between cysts. This classification holds for both ADPKD and PLD.
Treatment
In ADPKD, longevity is more related to kidney function than liver function, and renal failure is common, so that these patients end up on dialysis sooner or later. Liver function is seldom affected in these conditions, presumably because regeneration preserves functional liver volume. Treatment of the liver cysts, therefore, is directed at symptomatic relief from pressure symptoms. Patients with type I cystic disease are most easily treated because they have a relatively small number of large and easily discernible cysts. Type II is also amenable to surgical treatment because ablation of a few large cysts may provide adequate relief of symptoms. However, type III, in which there are innumerable small and medium cysts, is less amenable to ablation of the cysts. These patients may receive benefit, albeit transient, from partial hepatectomy.3 Therapy with somatostatin analogues theoretically could reduce the accumulation of cyst fluid, but the therapeutic use of, for example octreotide, has been met with only modest reduction in cyst volume (about 5%).4
Practice Point.
Patients with polycystic liver diseases do not develop liver failure but may develop portal hypertension.
Liver transplantation has also been performed for these patients, although it is important to weigh up the risks of surgery and long-term immunosuppression against the severity of symptoms. The cysts can be massive and multiple, so that symptoms cannot realistically be attributed to any specific cyst or cysts. However, destruction of the largest cysts can be associated with temporary relief of symptoms. This can be accomplished at laparotomy, laparoscopy, or percutaneously. At laparotomy or laparoscopy cysts can be de-roofed and allowed to drain into the peritoneum. Since the fluid is similar to serum it is rapidly re-absorbed, although ascites has been described. The cysts can also be drained percutaneously and a sclerosant introduced to destroy the secretory epithelium and hopefully prevent recurrence of the cyst. Since liver function is preserved, liver transplantation is seldom necessary except when the size of the liver severely compromises the patient’s quality of life.
Treatment of symptomatic cysts can be surgical or by interventional radiology.
Infection of a cyst is rare, but serious, and has a mortality rate of about 2%. These require antibiotic and external drainage as for hepatic abscess.
Autosomal Recessive Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease/congenital hepatic fibrosis (ARPKD/CHF) is caused by mutations in the PKHD1 gene located on chromosome 6p12. PKHD1 is a relatively large gene containing 66 coding exons, which demonstrates a complex splicing pattern. The gene codes for the protein fibrocystin, a hepatocyte growth factor receptor-like protein that functions on the primary cilia of renal and biliary epithelial cells (Fig. 25.1). Dysfunction of fibrocystin leads to abnormal ciliary signaling, which is normally required for regulation of proliferation and differentiation of renal and biliary epithelial cells.
This is a rare condition that occurs in about 1 : 40 000 live births. Its primary manifestation is renal cystic disease, with progressive renal failure. However, it is associated with disease in other organs, including chronic lung disease, growth retardation, intracranial aneurysms, adrenal insufficiency, and hypertension. The hepatic manifestations of this condition are Caroli disease5 and CHF. When both Caroli disease and CHF are present the condition is known as Caroli syndrome.
Caroli disease is characterized by multiple fusiform or cystic dilatations of the intrahepatic bile ducts that communicate with the biliary system. The major complications are related to recurrent bouts of cholangitis, and biliary stone formation. There is a risk of developing cholangiocarcinoma. In CHF, the portal tracts are abnormal, with multiple abnormally shaped embryonic bile ducts, often retaining the ductal plate conformation. The portal vein is abnormal. There is significant periportal and bridging fibrosis. The clinical manifestations are those of portal hypertension, including splenomegaly, thrombocytopenia, and variceal bleeding (and patients are thus frequently misdiagnosed as having cirrhosis). What they really have is “congenital hepatic fibrosis”. They may also have had medullary sponge kidneys and nephrocalcinosis diagnosed in the past.
These patients often present in utero, with oligohydramnios, but when they present later in life the symptoms are related to the liver and/or kidney disease. About 30% of cases die in the perinatal period with respiratory difficulty. Generally, liver cysts are not observed in children, but develop over time.
There are four variants of Caroli syndrome, with slightly differing clinical presentations. These are the portal hypertensive presentation, a cholangitic presentation with multiple episodes of cholangitis, a mixed picture, and a latent pattern, in which presentation occurs late in life, often presenting with complications, such as cholangiocarcinoma. On examination the liver is firm and enlarged. The spleen and kidneys may be enlarged. It may be difficult to tell whether the organ being palpated is liver and spleen or the kidneys. In those with a predominantly cholangitic or mixed picture, the alkaline phosphatase and gamma glutamyl transpeptidase are usually markedly elevated. The bilirubin may also be elevated, particularly during episodes of acute cholangitis.
Diagnosis
The disease is usually first detected, often incidentally, by ultrasonography. This might show, in addition to the cysts, changes in the sizes of various liver lobes or segments, focal or more generalized intrahepatic biliary dilatation and stones (Caroli disease), or splenomegaly. There may be periportal thickening (congenital hepatic fibrosis), cavernous transformation of the portal vein, and enlarged cystic kidneys. Magnetic resonance imaging (MRI) (Fig. 25.3) or magnetic resonance cholangiopancreatography (MRCP) have been used to evaluate the liver and biliary tree in the setting of ARPKD/CHF.6 Intrahepatic biliary ductal dilatation can be demonstrated on axial, coronal, and reformatted MR cholangiography images. A distinctive feature of CHF MR cholangiograms is that the ducts do not taper to the periphery of the liver as they do normally. Instead, they remain dilated throughout the parenchyma, often forming cisterns and small cysts at the periphery of the liver. MR cholangiography is also useful for detecting biliary system complications including stones and cholangitis and cholangiocarcinoma.
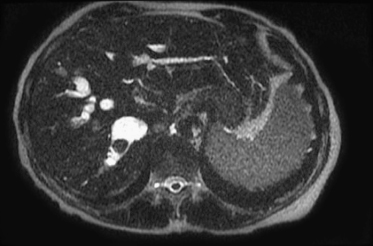
Fig. 25.3 Caroli disease. Note the fusiform dilatations of the biliary system, with a large stone present.
Related posts:
 9 Indications for liver transplantation in adults and children
9 Indications for liver transplantation in adults and children
 16 Drug-induced liver disease
24 Liver disease in pregnant women
16 Drug-induced liver disease
24 Liver disease in pregnant women
 14 Chronic viral hepatitis in adults and children: hepatitis C
14 Chronic viral hepatitis in adults and children: hepatitis C
 12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
 26 An internist’s approach to radiologic examination of the liver
26 An internist’s approach to radiologic examination of the liver
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


