- Wilson disease (WD) is a rare, but treatable, autosomal recessive disorder of hepatic copper disposition.
- Onset of clinical disease can occur at any age, though mainly between 3 and 55 years old. Age alone is not a reason for discounting the diagnosis of WD.
- WD can present as hepatic disease, neurological movement disorders, or psychiatric disease.
- In a patient with liver disease or typical neurologic features, the combination of subnormal serum ceruloplasmin (preferably <140 mg/dL) and elevated basal 24-h urinary copper excretion (>0.6 µmol/24 h or >40 µg/24 h) is highly suggestive of WD.
- Genetic diagnosis is definitive but requires skilled interpretation.
- Treatment is lifelong. Early diagnosis and treatment provide the best outlook. Discontinuing treatment altogether leads to severe refractory liver dysfunction; treatment should not be stopped during pregnancy.
- First-degree relatives must be investigated for WD once a single family member has been diagnosed with WD; genetic testing is most efficient.
Definition
Wilson disease is a genetic disorder of hepatic copper disposition. The abnormal gene is ATP7B, identified in 1993. The transmission pattern is autosomal recessive. The vast majority of affected individuals are compound heterozygotes.
Pathogenesis
ATP7B encodes the Wilson ATPase, a metal-transporting P-type ATPase, which has two principal functions in the hepatocyte. The Wilson ATPase is essential for incorporation of copper into nascent ceruloplasmin, and it expedites biliary excretion of copper. Thus, when the Wilson ATPase is absent or non-functional because of mutations in ATP7B, copper accumulates in hepatocytes. One reason for the copper accumulation is that excess copper is not excreted into the bile, and another is that copper is not used for producing ceruloplasmin. Ceruloplasmin is thus released into the bloodstream without containing copper. Since most of the copper measured in the blood is actually within ceruloplasmin, serum copper measurements are low in WD. Given the hepatic copper overload, this finding seems paradoxical. If copper is released from the liver, for example with progressive liver injury for fulminant hepatic failure, the serum copper is elevated. The excess copper in the plasma compartment is not “free” copper; instead it is loosely bound to albumin and certain amino acids.
As WD progresses, small amounts of copper leach out of the liver and accumulate in other organs: the brain, renal tubules, heart, and synovia, among others. Copper accumulating in Desçemet’s membrane of the cornea produces the Kayser–Fleischer ring. Self-limited episodes of copper released from the liver can cause transient hemolysis, resulting in mild jaundice.
Epidemiology
WD is found worldwide. The average prevalence is 30 affected persons per million. The corresponding carrier frequency is approximately 1 in 90. More than 500 mutations have been identified. No one single mutation is extremely common. Certain mutations are typical for individuals of specific ethnic or geographical backgrounds. In north-eastern Europe, H1069Q is found as at least one of the mutations in 35–75% of WD patients. A circumscribed set of mutations is found in certain other populations including Iceland, Korea, Japan, Sardinia, and the Canary Islands.
Clinical Features
WD can present clinically as liver disease, neurological disease usually with a movement disorder of some kind, or psychiatric disease.1 Some presymptomatic patients are identified through family screening once a proband has been diagnosed. Presymptomatic infants may be identified through universal newborn screening in countries where such programs are established. Hepatic disease is more common in younger patients and neuropsychiatric disease in somewhat older patients, but the demarcation is vague. Certainly, children do present with mainly neurological disturbances in WD.
The first clinical presentation of WD can occur at any age. Age at diagnosis ranges from approximately 4 to 70 years. Toddlers (1.5–3 years old) and seniors (>70 years old) have been diagnosed with WD.
Hepatic WD.
Hepatic involvement takes numerous forms. The patient may feel well but have moderately abnormal liver biochemistries. Non-specific symptoms such as fatigue or anorexia may be present. Hepatic steatosis (fatty liver, typically found by liver sonography) is common in WD, affecting up to half of patients. Some patients present with an illness closely resembling autoimmune hepatitis, and a few patients have a self-limited clinical illness resembling acute hepatitis with elevated serum IgG and presence of non-specific autoantibodies. Many patients present with cirrhosis and portal hypertension: hepatosplenomegaly, ascites, low serum albumin, and coagulopathy. These patients must be distinguished from those who present with fulminant hepatic failure (WD–FHF). Features of WD–FHF include acute Coombs-negative intravascular hemolysis, rapid onset of renal failure, disproportionately low serum aminotransferases (usually  1500 IU/L) from the onset of clinically apparent disease, subnormal or just normal serum alkaline phosphatase, in addition to severe coagulopathy and some degree of encephalopathy. WD–FHF is somewhat more common in females than males.
1500 IU/L) from the onset of clinically apparent disease, subnormal or just normal serum alkaline phosphatase, in addition to severe coagulopathy and some degree of encephalopathy. WD–FHF is somewhat more common in females than males.
Neurologic WD.
Patients with neurologic WD generally have some liver involvement although it may be asymptomatic. The most common clinical symptoms are speech defects, drooling, hand tremors, and gait disturbance.2 Overall, neurologic WD follows two broad patterns: movement disorders or rigid dystonia. Movement disorders typically occur in younger patients and include tremors, poor coordination, and loss of fine motor control. Tremor resembling a familial tremor is often present. Spastic dystonic disorders resemble Parkinson disease with mask-like facies, rigidity, and gait disturbance. Pseudobulbar involvement, such as dysarthria, drooling, and swallowing disorder, is more common in older patients but may occur in children and adolescents. Intellect is normal.
Psychiatric WD.
Personality changes may be apparent in patients with neurological WD, but an actual psychiatric disorder is present in the approximately 20% of WD patients who present with psychiatric WD. The spectrum of disorder is highly variable.3 Depression is probably most common. Neurotic behaviors such as obsessive–compulsive behaviors or phobias have been reported; aggressive or antisocial behavior may occur. Affected patients tend to be older and the psychopathology may be subtle: progressive disorganization of personality with anxiety and affective changes such as labile moods and disinhibition. Psychotic disorders are uncommon.
Associated Disorders
WD can affect numerous organ systems besides the liver and brain. These are summarized in Table 23.1.
Table 23.1 Systems affected by Wilson disease
| Liver | Fatty liver; resembling autoimmune hepatitis; cirrhosis, etc.; bilirubinate gallstones |
| Brain | Movement disorders resembling chorea; rigid dystonia; dysarthria; dysautonomia |
| Psychiatric manifestations | |
| Eyes | Kayser–Fleischer rings; sunflower cataract |
| Blood | Self-limited intravascular hemolysis |
| Kidney | Renal tubular dysfunction; Fanconi syndrome; kidney stones; hypokalemic muscle weakness |
| Joints | Arthralgia or arthritis; osteoporosis, osteopenia |
| Pancreas | Pancreatitis |
| Heart | Arrhythmias; cardiomyopathy |
| Endocrine | Hypoparathyroidism Testicular dysfunction Amenorrhea; spontaneous abortion; infertility |
The classic Kayser–Fleischer (KF) ring is due to copper accumulation in Desçemet’s membrane. Well-developed KF rings are a golden brown color and can be seen by direct inspection only if the iris is a pale color: thus slit-lamp examination is required. It is absent in half of the patients with hepatic WD and occasionally is absent in patients with neurologic WD. Although KF rings are generally specific for WD in children, they are not specific for WD in older patients. Copper deposited in the lens produces the sunflower cataract, which does not interfere with vision.
Hepatocellular carcinoma was thought to be exceedingly uncommon in WD, but accumulating data suggest that this is not the case. Hepatic and abdominal malignancies may be found in older patients despite adequate treatment.
Differential Diagnosis
The patient who presents with obvious liver disease (or typical neurological disorder), exceedingly low serum ceruloplasmin, and KF rings is easy to diagnose as having WD. Unfortunately, these patients are quite uncommon. A high index of suspicion is valuable for diagnosing Wilson disease. The differential diagnosis for WD falls into three major categories: hepatic disorders, neurological disorders, and diseases associated with low serum ceruloplasmin. The latter include protein-losing enteropathy, nephrotic syndrome, and malnutrition, and the rare genetic condition aceruloplasminemia associated with neurological, retinal, and pancreatic degeneration due to iron accumulation in the brain, retina, and pancreas.
Evaluation of Liver Disease
Severity of liver disease needs to be determined. Many patients have comparatively mild liver disease without much scarring or vascular damage. Those who have decompensated cirrhosis with coagulopathy, low serum albumin, and some degree of jaundice may still respond to medical treatment. Patients with WD–FHF or treatment-refractory liver disease require liver transplantation.
Presence of any liver disease must be ascertained in the first-degree relatives of a newly diagnosed patient with WD. A clinical strategy for such investigations is given in Table 23.2. Genetic testing is the most efficient way to diagnose WD in first-degree relatives, if the proband’s genotype is known. If a brother or sister is a potential donor for a living donor liver transplant, genotypic diagnosis is essential.
Table 23.2 Strategy for investigation of first-degree relatives of a person newly diagnosed with Wilson disease (WD)
| Molecular testing available | No molecular testing available | |
| Brother or sister of patient | Haplotype or mutation analysis Identical haplotype or two mutations → WD: baseline testing then treatment Different haplotype or no mutations → diagnosis excluded Indeterminate, such as only one mutation found → baseline testing | Baseline testing Serum for liver biochemistries, ceruloplasmin, INR, complete blood count Urine for 24-h urinary copper excretion (basal) Slit-lamp examination |
| All normal → diagnosis excluded Abnormal liver tests or subnormal ceruloplasmin or elevated 24-h urinary copper excretion (>0.6 µmol/24 h) → liver biopsy with measurement of hepatic copper: Hepatic copper <50 µg/g dry weight → diagnosis excluded Hepatic copper >250 µg/g dry weight → WD, treatment Hepatic copper 51–250 µg/g dry weight → get molecular testing | ||
| Child of patient (if free of symptoms, test at >2 years old) | Haplotype or mutation analysis Identical haplotype or two mutations → WD, baseline testing then treatment Different haplotype or no mutations → diagnosis excluded Indeterminate, such as only one mutation found → baseline testing (no slit lamp exam until child >4 years old) | Baseline testing (as above) All normal → diagnosis excluded Abnormal liver tests → repeat baseline testing in 6–12 months Subnormal ceruloplasmin or elevated 24-h urinary copper excretion (>0.6 µmol/24 h) → liver biopsy with measurement of hepatic copper or get molecular testing |
Laboratory Features
Routine liver biochemistries are usually abnormal with mild to moderately elevated serum aminotransferases. Biochemical testing may miss very early disease. Serum aspartate aminotransferase (AST) may be higher than alanine aminotransferase (ALT) because of mitochondrial damage or hemolysis. With more advanced disease, serum albumin may be low. Since serum albumin is normal in true acute liver failure, hypoalbuminemia may be an additional clue to WD–FHF. Mild coagulopathy may be the last laboratory feature to improve on medical treatment.
Serum ceruloplasmin is usually subnormal. Approximately one-third of patients have serum ceruloplasmin below 100 mg/L; one-third have serum ceruloplasmin between 100 and 200 mg/L; the remaining third have normal serum ceruloplasmin. Ceruloplasmin by itself is not adequate for diagnosing WD and is of questionable value in screening for WD. The main reason for the inadequacy of ceruloplasmin as a diagnostic criterion now, as compared to 50 years ago, is that the automated test is performed by immunological, not enzymatic, methods. Thus it does not determine whether copper-containing ceruloplasmin is present. Recent studies suggest that in adults serum ceruloplasmin below 140 mg/L is specific and sensitive for the diagnosis of WD.4
In WD serum copper concentration is low, in parallel with the low serum ceruloplasmin. The non-ceruloplasmin-bound copper concentration is elevated. Although direct methods for measuring non-ceruloplasmin-bound copper are bring developed, this fraction is usually estimated from the proportion of copper known to be associated with ceruloplasmin. The estimate depends on the accuracy of the ceruloplasmin measurement and has not been validated as a diagnostic tool. In normal individuals, the non-ceruloplasmin-bound copper is approximately 50–100 µg/L. In WD, the non-ceruloplasmin copper is more than 200 µg/L, but it may be even 10 times higher in WD–FHF.
Measurement of basal 24-h urinary excretion of copper is highly informative for the diagnosis of WD. Recent studies show that the cut-off for suspecting WD should be 0.6 µmol/24 h (>40 µg/24 h). Using the higher conventional reference values severely limits the utility of this test. The penicillamine challenge test has proven less reliable than previously thought, and it may add no extra information beyond what can be gained through measuring basal excretion of copper, which reflects the total body copper load and, more directly, the non-ceruloplasmin-bound copper.
Hepatic tissue copper concentration is highly informative in most WD patients. Hepatic copper content greater than 250 µg per gram dry weight is accepted as diagnostic of Wilson disease; however, recent data indicate that 100 µg per gram dry weight might be a better diagnostic threshold. Indeed, the hepatic copper concentration is generally 10 or more times the value found in normal individuals. Providing a large enough sample of liver tissue is important for accurate measurement. With cirrhosis, copper may be variably accumulated in different nodules. Some heterozygotes have moderate elevations of liver tissue copper, without having clinical liver disease. Finding elevated hepatic copper concentration also is not specific for WD; patients with chronic cholestasis or diseases such as Indian childhood cirrhosis may have elevated hepatic copper. Liver copper can be measured in formalin-fixed specimens.
For WD–FHF a simple formula of routine laboratory tests has proven sensitive and specific: alkaline phosphatase/total bilirubin < 4 plus AST/ALT > 2.2, all in American units.5 For WD in general, a diagnostic profile has been developed but remains incompletely validated; moreover, definite limitations to its diagnostic accuracy have been identified.
Investigations
Imaging the liver may show fatty liver, nodular transformation, or features suggestive of cirrhosis. Imaging the central nervous system may be informative and should be performed even in the absence of neurological disorder. Magnetic resonance imaging (MRI) is more sensitive than computed tomography (CT). Generalized cerebral atrophy may be present. Abnormalities of the putamen and pons are most frequently found in patients with a neurological presentation; in patients with an hepatic presentation abnormalities of the globus pallidus are common. Only the so-called “face of the giant panda” sign seems to be specific for neurological WD.
Liver biopsy may provide important diagnostic information besides tissue for copper quantification, and it permits staging of the liver disease.6 A routine disposable needle can be used; the specimen should be aliquoted immediately for histology and copper quantification with minimal exposure to metals. Earliest histological findings include steatosis, focal hepatocellular necrosis, and/or apoptotic bodies, and glycogenated nuclei in hepatocytes. Mallory–Denk bodies may be found. With progressive parenchymal damage, possibly by repeated episodes of lobular necrosis, periportal fibrosis develops. Typically, cirrhosis is macronodular. Commonly used histochemical stains for copper may be negative in the early stages of WD because only copper aggregated in lysosomes is stained. On electron microscopy (EM), classic changes in mitochondria may be identified. Mitochondria vary in size, with increased matrix density and dark inclusions; however, the most striking change is dilatation of the tips of the mitochondrial cristae so that each crista appears cystic, shaped like a tennis racquet rather than a shelf. Mitochondrial changes occur early in WD. If WD is the leading possible diagnosis, a portion of the biopsy should be placed in universal fixative immediately for possible EM examination.
Genetic diagnosis is definitive. The gene ATP7B is found on 13q14.3. It is a large gene. Mutations tend to be missense, nonsense, and splice-site mutations, and large deletions are uncommon. Mutations that interfere with production of the Wilson ATPase cause severe liver disease,7 but extensive genotype–phenotype correlations have been elusive, mainly because most WD patients are compound heterozygotes with one copy of two different mutations. Modern high-throughput analytic methods make it possible to analyze the entire gene fairly easily; haplotype analysis and other methods are also available. A small percentage of patients who have WD by clinical diagnosis nevertheless appear to have no detectable genetic abnormality, and current research is aimed at understanding genetic mechanisms in these patients. Primary genetic diagnosis may not yet function as the most efficient diagnostic investigation, but it is ideal for evaluating siblings.
Natural History
Untreated WD is fatal. It has a good prognosis if it is diagnosed promptly and treated effectively. Treatment is lifelong. The best outlook is for the person with presymptomatic WD who starts treatment before any clinical abnormality is apparent.8 Patients presenting with early hepatic disease generally have a good prognosis so long as treatment is taken consistently and is well tolerated, but some patients with hepatic WD develop progressive liver disease despite treatment.2 Patients who discontinue treatment entirely tend to develop refractory liver disease or new neurological involvement. Women who discontinue their medication for WD during pregnancy may develop severe hepatic deterioration or liver failure postpartum. Severe neurological disease may not resolve entirely on drug treatment, although substantial improvement usually occurs.9
Liver transplantation should be reserved for patients who present with severe, decompensated liver disease unresponsive to medical therapy or for those who present with WD–FHF. After liver transplant, chelation therapy is no longer required. The efficacy of liver transplantation to improve neurological WD is highly disputed, and on balance it appears ineffective. Some WD patients have significant difficulty complying with the medical regimen after liver transplantation.
Patients with WD need to make a concerted effort to maintain good general health. Being overweight or obese should be avoided. Because of the potential for further hepatic mitochondrial damage, they should avoid—or at least limit—alcohol. Like any patient with chronic liver disease, they should have vaccinations for hepatitis A and B. Since WD may predispose to osteoporosis, all patients need to be assessed for osteoporosis and they should adopt lifestyle habits that promote maintenance of healthy bones.
Therapy
Therapy, summarized in Table 23.3, is guided mainly by clinical experience as few randomized controlled trials have been performed.1,10 Large retrospective studies have recently become available. Some clinical studies investigating one treatment or another are subject to selection bias. Heterozygotes do not require treatment.
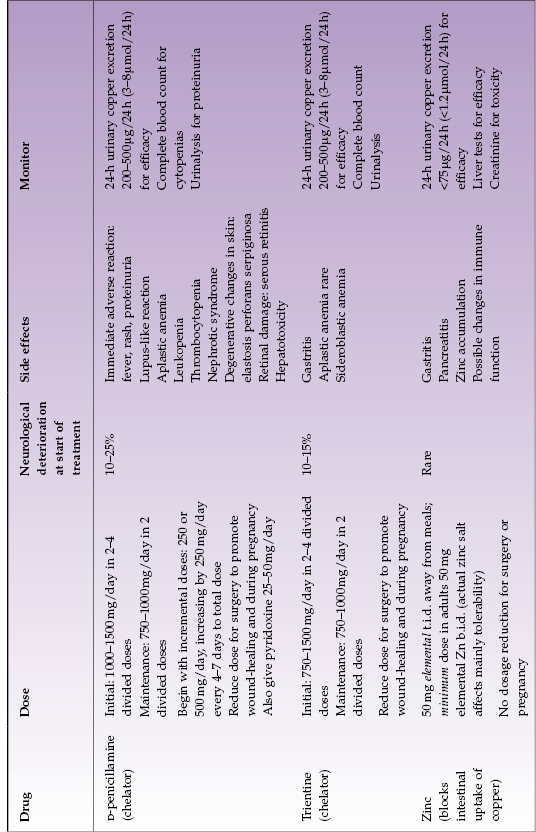
Table 23.3 Treatment options for adults with Wilson disease (see text for children’s treatment)
D-Penicillamine.
The first effective oral treatment of WD was introduced in the 1950s: D-penicillamine, an oral chelator. It markedly increases urinary excretion of copper and induces hepatic metallothioneins; additionally, it inhibits collagen cross-linking and has some immunosuppressant properties. It has proven to be very effective for treating WD. Unfortunately, it has important adverse side effects. Approximately 10–25% of patients with neurologic WD have a deterioration of their neurological status when they start D-penicillamine. Various types of skin disorders can occur, including rashes, pemphigus, and elastosis perforans serpiginosa. Loss of taste, althralgias, and diarrhea have been reported. The very severe side effects of D-penicillamine include proteinuria, leukopenia, or thrombocytopenia. Aplastic anemia is a rare catastrophic complication, which does not always reverse when D-penicillamine is stopped. Nephrotic syndrome, systemic disease resembling lupus erythematosus, Goodpasture syndrome, and a myasthenia gravis-like syndrome have all been reported in patients receiving chronic treatment with D-penicillamine. Severe side effects require immediate discontinuation of D-penicillamine with substitution of alternate therapy. Some patients develop an early adverse reaction with fever, rash, and proteinuria (within 7–10 days of beginning treatment); this mandates changing to another treatment, usually trientine.
The usual adult maintenance dose of D-penicillamine is 1000 mg daily, divided into two daily doses. Compliance and efficacy should be checked measuring 24-h urinary copper excretion every 6–12 months; complete blood count and urinalysis should be monitored regularly throughout its use. Pyridoxine 25 mg daily should also be given because of possible adverse effects on pyridoxine metabolism associated with D-penicillamine administration. Children’s dosing is 20 mg/kg as total daily dose plus pyridoxine.
Trientine.
This chelator has been used since the early 1980s and differs chemically from penicillamine by the absence of sulfhydryl groups. Thus it is a good substitute for D-penicillamine. It is highly effective primary therapy in children and adults, and it should be used as first-line treatment whenever there is apparent predisposition to adverse effects of D-penicillamine. Trientine is well-tolerated in WD apart from occasional gastritis. It can cause iron-deficiency anemia, apparently by chelating dietary iron, or rarely sideroblastic anemia. Bone marrow suppression is extremely rare. Neurological worsening after beginning treatment with trientine has been reported in neurologic WD, but it appears less frequent than with D-penicillamine.
The usual total daily dose is 1000–1200 mg (depending on tablet size available) divided into two daily doses, preferably taken not at mealtime (for example, the typical regimen is before breakfast and at bedtime). Monitoring for effectiveness and safety is as for penicillamine (notably, measuring 24-hour urinary copper excretion every 6–12 months and checking the complete blood count). Children’s dosing is 20 mg/kg as total daily dose.
Zinc.
Zinc salts have been utilized for treating WD since the 1960s (Europe) and the 1980s (North America). In pharmacological doses, zinc interferes with absorption of copper from the gastrointestinal tract by inducing enterocyte metallothionein, which has greater affinity for copper than for zinc and thus preferentially binds copper present in the intestinal contents. Once it is bound, the copper does not get absorbed and it is lost in the feces due to normal turnover of enterocytes. Since absorbable copper enters the gastrointestinal tract from the diet and from internal secretions (except bile), it is likely that zinc treatment mobilizes endogenous copper. The actual zinc salt does not matter to the mechanism of action but may affect tolerability. The common side effect of gastritis can be minimized by using a salt other than sulfate. In general, zinc has few adverse side effects. Neurological deterioration is uncommon.11 Zinc treatment has been well tolerated in adults and children. Recent data suggest that zinc is not entirely effective for controlling hepatic WD over the long term. A disadvantage of zinc is that t.i.d. dosing is required and zinc should be taken away from meals; these dosing requirements may make adherence to the regimen difficult.
The usual adult daily dose is 50 mg elemental zinc taken three times a day away from meals. Single daily dosing is ineffective; b.i.d. dosing may not be effective. Children’s dose is 25 mg elemental zinc taken three times a day away from meals.
Tetrathiomolybdate (TTM).
Ammonium tetrathiomolybdate remains an investigational drug especially suitable for treatment of severe neurological Wilson disease because, unlike D-penicillamine, it is not associated with early neurological deterioration. It is a strong chelator. Adverse effects have been reported including overly aggressive removal of copper, and the disposition of administered molybdenum is not entirely clear. TTM is not generally available for clinical use.
“Combination Treatment”.
Currently there is much interest in combining a chelator with zinc in order to maximize therapeutic effect synergistically. Either D-penicillamine or trientine is combined with zinc.12 This ends up as a q.i.d. regimen. The two types of treatment must be temporally dispersed throughout the day, with usually 5–6 hours between administration of either drug, or else there is the likelihood that the chelator binds the zinc and thus negates the efficacy of both therapeutic modalities. A typical, possibly preferred, regimen is zinc (50 mg elemental, or 25 mg elemental in children) given by mouth as the first and third doses, and trientine (500 mg, or 10 mg/kg in children) given by mouth as the second and fourth doses. This strategy is fundamentally an induction regimen suitable for patients who present with severe disease. Some will fail and require liver transplantation, which should be arranged as a back-up. If it is effective, then it should be continued for 3–6 months after which time the clinically stable patient can be transitioned to monotherapy, preferably with the chelator. Long-term adherence to this intensive regimen appears neither necessary nor practical.
Antioxidants.
Since oxidant stress plays an important role in the disease mechanism of hepatic WD, antioxidants might be effective. Based on anecdotal evidence, at a dose of 400–800 IU daily, natural-source vitamin E may be useful adjunctive therapy.
Diet.
WD cannot be treated by dietary intervention alone. Especially in the first year of treatment, copper-rich foods should be removed from the diet. These foods include organ meats, shellfish, nuts, chocolate, and mushrooms. In real life, this means eliminating chocolate, nuts, and shrimp. Vegetarians require formal dietary counseling because legumes are high in copper. Installing a device in the plumbing system to adsorb copper is necessary only if the drinking water has a high copper concentration, possible with well water, or the plumbing has been installed with copper pipes.
Liver Transplantation.
With early diagnosis and effective treatment, liver transplantation can usually be avoided in WD. Patients who do not respond to treatment or who present in WD–FHF require liver transplantation. It corrects the hepatic metabolic abnormalities and may improve extrahepatic copper handling. One-year survival is on the order of 80–90%, and most who survive the first year do well long term.13 Living-donor transplantation is effective even if the donor is a heterozygote.14 With WD–FHF, plasmapheresis, exchange transfusion, or albumin dialysis may stabilize the patient and minimize renal damage. These interventions extend the time available for actually obtaining a liver graft and, very rarely, may obviate the need for transplantation.
Special Therapeutic Challenges
Decompensated Chronic Liver Disease.
These patients may be diagnosed as having acute liver failure, especially if the criterion of encephalopathy is discounted. Many of these patients will respond to an intensive medical regimen, such as the combination regimen described above. Automatic liver transplantation may be inappropriate, although liver transplantation should be available as an urgent alternative and thus a consultation should be requested.
Pregnancy.
Treatment must not ever be stopped just because a woman is pregnant; liver failure may develop. D-penicillamine is classified as a teratogen. Treatment can be converted to trientine or zinc for the duration of the pregnancy. Insufficient data are available to judge the safety of these drugs in pregnancy. The dose of either chelator should be decreased in the third trimester by 25–50%, but the dose of zinc need not be altered. Women with extensive liver disease should receive obstetric care by a team in a high-risk pregnancy program. With patients on D-penicillamine, breast feeding should be discouraged; little is known about the safety of either trientine or zinc for breast feeding.
Infants with WD Identified by Universal Newborn Screening.
Copper is required for normal development of the central nervous system. The possible adverse effects of zinc overload on development are not known. Serum ceruloplasmin is physiologically low in early infancy. At the present time there is no standard regimen for treating infants with presymptomatic WD. Treatment with zinc is favored, perhaps starting when the child is a toddler. Very young children with clinically evident WD require treatment with a chelator.
Presymptomatic Disease.
Most of these patients are best treated with a chelator. For the patient, identified by family screening, who has absolutely no abnormalities (liver tests normal, imaging normal), zinc may be preferred; however, t.i.d. dosing may prove problematic.
Stable Patients on Chelator Therapy.
Zinc has a role in long-term maintenance therapy, although the specifics of that role are still being determined. Switching over to zinc can be considered in a patient who is clinically stable on a chelator after 1–5 years (typically 3–5 years) of treatment and has an excellent record of adherence to medical regimen. Liver biochemistries should be normal, as well as copper homeostasis. Close follow-up is still required, and if liver tests become abnormal, the chelator should be restarted. No patient with proven WD should be permitted to stop all treatment completely.
Related posts:
 9 Indications for liver transplantation in adults and children
9 Indications for liver transplantation in adults and children
 16 Drug-induced liver disease
24 Liver disease in pregnant women
16 Drug-induced liver disease
24 Liver disease in pregnant women
 14 Chronic viral hepatitis in adults and children: hepatitis C
14 Chronic viral hepatitis in adults and children: hepatitis C
 12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
 26 An internist’s approach to radiologic examination of the liver
26 An internist’s approach to radiologic examination of the liver



