- Metabolic liver diseases have a wide clinical spectrum of presentation and are often asymptomatic in adults. A high index of suspicion must be maintained in order to achieve a diagnosis.
- In children and adults, metabolic disease is often insidious, progressing silently to cirrhosis and subsequent liver failure.
- There have been many therapeutic advances for the metabolic liver diseases but good outcome depends upon early detection.
- In the pediatric population, enzymatic deficiencies of the essential metabolic pathways cause severe liver disease with or without neurologic disease.
- Diagnosis in the pediatric population usually requires direct enzyme analysis or genetic testing.
- Liver transplantation corrects only the hepatic phenotype abnormality in metabolic diseases due to enzymatic deficiency, but often this is sufficient for generally improved health.
The liver has numerous metabolic functions. Metabolic liver disease includes a wide variety of clinical disorders and pathophysiology (Table 22.1). Most are due to inherited enzymatic abnormalities, which result in storage disorders, errors of metabolism, and transport defects. These diseases can manifest as acute and life-threatening illnesses in the neonatal period or as chronic liver illness in adolescence or adulthood. In the pediatric population, up to 20% of liver transplants are performed for complications resulting from a metabolic liver disease whereas in the adult population it is approximately 5%. Metabolic liver diseases are great mimics. For example, citrullinemia type 2 (discussed here) and Wilson disease (Chapter 23) can both resemble non-alcoholic fatty liver disease (NAFLD; Chapter 17). To diagnose metabolic disease, a structured approach is required, which includes consideration of the age at presentation, symptoms, laboratory abnormalities, and specific diagnostic testing.
Table 22.1 Selected metabolic diseases with mainly adult or child/adolescent presentation
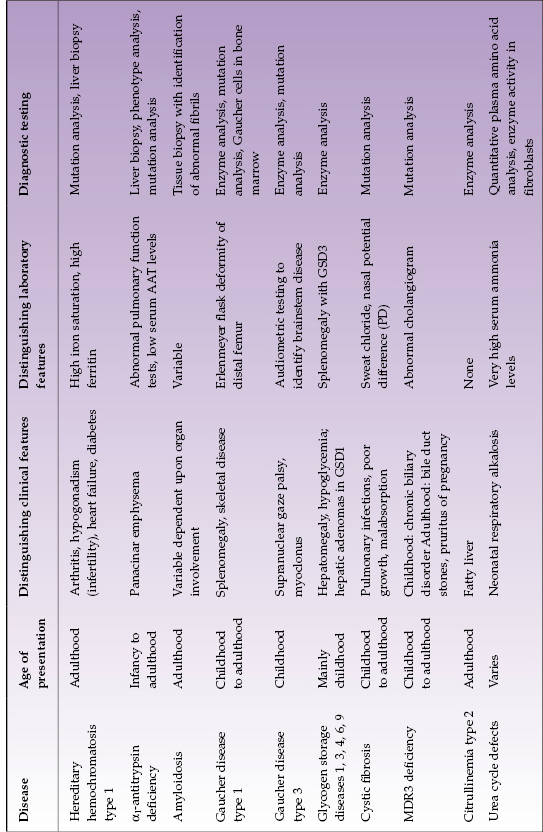
Hereditary Hemochromatosis
Hemochromatosis is a clinical syndrome of iron overload, which may be due to genetic abnormalities or to exogenous iron overload by transfusion or food contamination. Hereditary hemochromatosis (HH) comprises several inherited disorders of iron homeostasis, all of which lead to increased intestinal iron absorption and tissue iron deposition. Classic (type 1) HH is the well-known form of iron overload due to HFE1 (“high Fe”) gene defects. It affects 0.5% of Caucasians and currently is most often detected during the work-up for arthritis or infertility or other disease (such as heart failure or diabetes) with biochemical markers of increased iron stores. It should be considered in all Caucasians with unexplained abnormal liver tests (Fig. 22.1). Several other genetic disorders of iron metabolism that cause HH have been identified but are much less common (Table 22.2).
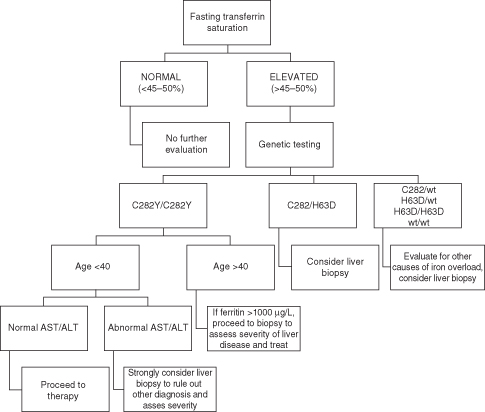
Table 22.2 Classification of hemochromatosis
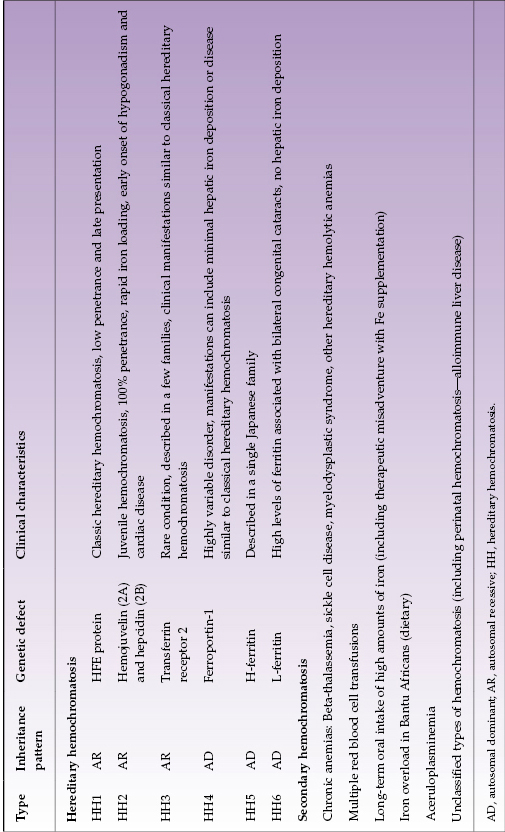
Type 1 HH is the most prevalent inherited metabolic disease in Caucasians. The only important mutations of the HFE1 gene are C282Y and H63D. Both C282Y/C282Y homozygotes and C282Y/H63D compound heterozygotes can manifest disease, although penetrance is markedly higher in homozygotes than compound heterozygotes. C282Y homozygotes account for 90% of type 1 HH. Phenotypic expression of disease depends upon non-genetic factors such as amount of iron consumption and/or blood loss; for example, women develop clinical consequences of iron overload four to eight times less frequently than men and 10–20 years later due to menstruation.
HFE1 gene defects lead to increased intestinal iron absorption. Usually, only 10% of daily iron intake (1–2 mg) is absorbed in the small bowel. However, individuals with C282Y homozygosity may absorb up to 40% of their daily iron intake (4–8 mg). The mechanism by which this occurs is through the circulating peptide hormone, hepcidin. In normal patients, hepcidin is produced in the liver and increases in response to increased levels of circulating iron, signaling to intestinal cells to decrease iron absorption. However, in patients with classic hereditary hemochromatosis, hepcidin production in the liver is impaired and leads to uncontrolled intestinal iron absorption. Organ damage occurs when iron stores approach 10 g and thus it may take many years before clinical end-organ damage is detected. Patients with clinical end-stages of disease typically have 20–30 g of iron stores.
Iron overload in the liver induces lipid peroxidation, and membrane-dependent functions of mitochondria, microsomes, and lysosomes become abnormal. There is a clear relationship between the degree of lipid peroxidation and hepatic fibrosis. As liver disease progresses, cirrhosis can develop. Late non-hepatic manifestations include hyperpigmented “bronzing” of skin, heart failure, hypogonadism, and diabetes.
Transferrin saturation is a more accurate indicator of iron overload than serum ferritin. High transferrin saturation usually precedes increases in ferritin. Transferrin saturation should be measured in the fasting state. A fasting transferrin saturation above 50% in women and above 60% in men will detect over 90% of patients with homozygous HH1. Ferritin is an acute phase reactant as well as an indicator of increased iron stores; low values can be found with iron deficiency and thus it used to monitor therapeutic response to therapy. In patients with elevated transferrin saturation, genetic testing for HFE gene mutations should be performed. Liver biopsy can be a useful adjunct to serum diagnostic testing because hepatic iron concentration can be measured and values higher than 71 µmol/g are highly suggestive of HH. For patients less than 40 years old, who are homozygous for C282Y and have no clinical features to suspect advanced fibrosis, biopsy is usually not indicated (Fig. 22.1).
Screening.
The rationale for screening for hemochromatosis is that treatment of asymptomatic disease prior to significant iron overload will prevent end-organ damage. Thus, screening (with iron saturation and/or HFE1 gene testing) is recommended for first-degree relatives of affected individuals. Population screening is not recommended.
Treatment.
Treatment for HH is mainly medical. The most uncomplicated and cost-effective treatment for hemochromatosis in non-anemic patients with iron overload is therapeutic phlebotomy (Box 22.1). Each 500 mL of blood removed contains 200 to 250 mg of iron. Most patients (even those with established cirrhosis) with HH1 or any genetic hemochromatosis will benefit from therapeutic phlebotomy unless their life expectancy is short for other reasons. Phlebotomy itself can be performed at a blood bank or medical facility, and the blood that is removed can be safely transfused to other individuals (provided that it passes other screening tests). While the optimal schedule is patient-dependent, in general iron should be depleted as rapidly as possible. Most male patients will tolerate 1 to 2 units of blood removed per week, while others may tolerate less. The goal of therapy is to deplete total body iron stores, which is achieved when the ferritin level drops to between 50 and 100 µg/L or the patient develops iron deficiency anemia. It is not uncommon to need 70 to 100 phlebotomies to deplete iron stores adequately before a maintenance phlebotomy schedule is put in place.
- Begin with one phlebotomy (500 mL) once or twice per week
- Check hematocrit prior to each phlebotomy; hematocrit should not fall by more than 20% of prior level
- Check serum ferritin every 10 to 20 phlebotomies
- Reduce frequency of phlebotomies with ferritin 100 µg/L
- Continue ferritin every 1 to 6 months to keep ferritin at approximately 50 µg/L
Patients with hemochromatosis are often concerned about consuming excess iron in their diet. However, given that most ingested iron is not absorbed, avoiding iron-containing foods is unnecessary. The one caveat to this principle is that excess alcohol consumption should be avoided as it is significantly associated with increased iron absorption and progression of liver disease.
Iron chelation therapy is available but usually not needed for genetic hemochromatosis. It is most commonly used for secondary hemochromatosis in patients who receive chronic transfusion therapy for anemia. Iron chelation therapy can be cumbersome when it requires continuous subcutaneous or intravenous infusion. Deferoxamine is a widely used infusional agent. Side effects include visual loss and ototoxicity. However, oral agents are now available, which are effective in most conditions. Deferasirox is a commonly used oral agent.
Hemochromatosis is an uncommon indication for liver transplantation. It is only considered for patients with end-stage hepatic disease and preserved cardiac function. Hepatocellular carcinoma may develop in cirrhotic HH1.
α1-Antitrypsin Deficiency
α1-Antitrypsin (AAT) deficiency leads to an increased risk of both lung and liver disease. AAT is a protease inhibitor that binds to the serine proteases in serum and tissues and promotes their degradation. The most important of the serine proteases is elastase. Loss of serum AAT activity leads to uninhibited neutrophil elastase activity and is the primary mechanism for the development of emphysema. Lung disease is characterized by the development of panacinar emphysema and its presence is independent of liver manifestations. Liver disease presents as an inactive cirrhosis and is caused by structural misfolding and polymerization of mutant AAT in the liver, leading to its retention in the endoplasmic reticulum of hepatocytes. It is not known exactly how AAT retention leads to liver injury.
The pattern of inheritance is codominant. Variants of the AAT allele produce gene products that can be distinguished by electrophoresis. Protease inhibitor MM (PI MM) is the normal allelic representation. The PI Z variant produces a mutant protein and homozygosity of the allele (PI ZZ) is the most common form of AAT deficiency leading to liver disease. More than 70 naturally occurring allelic variants have been described, but only some lead to chronic liver failure and cirrhosis in adults. More than 90% of all AAT is produced in the liver, and each allele contributes to the total amount of AAT produced. Thus individuals with the PI MZ phenotype have approximately half as much AAT as normal, and those with PI SZ have less than half and may develop clinical liver disease. AAT is an acute phase reactant. Serum levels of AAT may be artificially elevated by hepatic inflammation, and therefore phenotyping is the reliable diagnostic method. Genetic testing is an option; the mutation responsible for the Z protein is lysine replacing glutamic acid at position 342.
Diagnosis of AAT deficiency should be made with phenotypic analysis of the enzyme. While AAT levels can be measured, it should be noted that AAT is an acute phase reactant and thus levels can rise in pregnancy, with stress, or injury, even in PI ZZ patients. A liver biopsy is not universally recommended but should confirm the diagnosis if performed. Histologically, in infants, the biopsy can show bile duct loss, intracellular cholestasis, and mild inflammatory changes, but few of the characteristic periodic acid–Schiff (PAS)-positive globules. In adults, PAS-positive, diastase-resistant globules are usually seen. The inclusions are most prominent in periportal hepatocytes. Cirrhosis may be present.
The prevalence of PI Z is highest in populations derived from northern European ancestry, though many racial subgroups may be affected. About 10% of newborns with PI ZZ present with conjugated hyperbilirubinemia, and up to 50% have elevated serum aminotransferase levels by age 3 years (though most are asymptomatic). In adults lung disease generally precedes the development of liver disease. Even though liver disease is generally mild during infancy, patients with AAT deficiency have an eightfold higher risk of developing cirrhosis during adulthood. Additionally, homozygous AAT deficiency increases the risk of hepatocellular carcinoma. The diagnosis of AAT deficiency should be considered in any patient presenting with signs of cirrhosis. The presence of PAS-positive globules in hepatocytes on liver biopsy is almost always due to AAT deficiency, and this staining should be performed routinely on all liver biopsies.
Medical treatment is limited to lung disease. Most medical therapies for AAT deficiency are targeted at restoring AAT levels in the lung. No effective medical therapy is currently available to prevent progressive liver damage, although drugs that promote proper protein folding are being investigated. Gene therapy to restore the normal function of AAT to deficient human cells requires more study. Liver transplantation is reserved for patients with end-stage hepatic disease. It may be performed in conjunction with lung transplantation. Transplantation of a liver from a PI MM individual restores the normal hepatic phenotype as donor livers will produce and secrete AAT.
Amyloidosis
Amyloidosis presents in a myriad of ways. By infiltration into organs it can cause renal failure, heart failure, gastrointestinal bleeding, motor and sensory neuropathy, muscular pseudohypertrophy, bleeding diathesis, tracheobronchial fistulization, and pleural effusions. In the liver, it typically presents as massive hepatomegaly, with or without splenomegaly, or as mild chronic hepatitis. Amyloidosis refers to the extracellular deposition of fibrils composed of low molecular weight subunits of a variety of circulating proteins.
Several types of amyloid are hereditary and result from missense mutations of precursor proteins. Virtually all of the hereditary forms of amyloid are codominantly inherited heterozygous disorders, where both wild type and mutant molecules are identified in the amyloid deposits. There are several types of amyloidosis. AL (primary) amyloidosis is due to deposition of protein derived from immunoglobulin light chain fragments. This condition is a plasma cell dyscrasia in which a monoclonal immunoglobulin is detectable in serum. It can occur alone but is often associated with multiple myeloma, Waldenström macroglobulinemia or non-Hodgkin lymphoma. AA (secondary) amyloidosis occurs as a complication of a chronic inflammatory disease such as rheumatoid arthritis, spondyloarthropathy, inflammatory bowel disease, chronic infections, or periodic fever syndromes. Dialysis-related amyloidosis is a result of deposition of β2-microglobulin fibrils found in long-term hemodialysis patients. Other types of amyloidosis include familial amyloid polyneuropathy, age-related amyloidosis, and organ-specific amyloidosis.
Diagnosis of amyloidosis requires tissue biopsy in the suspected involved organ. If hepatomegaly is present, the liver has a thick, rubbery feel to it. The sensitivity of a liver biopsy in systemic amyloid disease when hepatomegaly is present is approximately 90%. Of note, liver is not the preferred site for tissue sampling because these patients have slightly higher than normal postbiopsy bleeding rates (though liver biopsies are not contraindicated). Biopsies of abdominal fat or rectal tissue have high sensitivity. The amyloid deposits appear as amorphous hyaline material deposited around hepatocytes on light microscopy; with Congo red staining they have green birefringence under polarized light. In some cases, immunohistochemical staining can help differentiate the various types of amyloid.
Treatment of amyloidosis varies with the type of fibrils. AL amyloid is treated by management of the plasma cell dyscrasia. AA amyloid is treated by management of the underlying condition leading to fibril formation (i.e. optimized control of rheumatoid arthritis). For the hereditary amyloidoses where mutant protein is produced by the liver, early liver transplantation can prevent further fibril deposition and lead to long-term regression of deposits.
In particular, liver transplantation has shown promise for familial amyloid polyneuropathy when performed based upon the severity of the sensorimotor neuropathy.
Gaucher Disease
Gaucher disease (GD) is an error of metabolism that results from a deficiency of the lysosomal enzyme glucocerebrosidase, leading to deposition of glucosylceramide and other glycolipids in lipid-laden macrophages of the reticuloendothelial system. In the liver, pathologic accumulation of these lipid-laden macrophages leads to an inflammatory and hyperplastic cellular response. GD, an autosomal recessive disorder, is the most common lysosomal storage disease and is most prevalent in the Ashkenazi Jewish population. For affected individuals, testing of all first-degree relatives with enzymatic functional assays and gene sequencing is recommended.
GD involves the visceral organs, bone marrow, and bone in all affected persons. Presenting features are variable and can present at any age. There are three subtypes of GD. Hepatomegaly is a feature in all three subtypes, and approximately 60% of patients at presentation have a liver volume of two to three times normal.
Type 1 GD is characterized by variability in age of onset; while it can present in infancy, some patients have no symptoms until adulthood. In addition to hepatomegaly, splenomegaly is the most common presenting sign with a median spleen size of 15 times normal. While hepatic fibrosis occurs, cirrhosis and liver failure are uncommon (but do occur). Patients with type 1 GD also develop skeletal disease (bone pain, avascular necrosis, joint collapse, osteolytic lesions, pathologic fractures), growth and developmental problems, interstitial lung disease, peripheral polyneuropathy, and are at increased risk of hematologic malignancies. Ninety percent of all GD patients have type 1 disease.
Type 2 GD is characterized by early onset of disease, typically within the first year of life. The first sign of disease is usually central nervous system disease (such as oculomotor dysfunction). Visceral involvement is extensive and severe.
Type 3 GD typical presents in childhood as a neurologic disease and visceral organ involvement, and varies by subtype. Type 3b has severe visceral involvement (hepatomegaly, bone disease) whereas type 3c has little visceral disease.
Diagnosis. Diagnosis of GD is suspected after identification of “Gaucher cells” in the blood; Gaucher cells are macrophages filled with lipid material. Other laboratory findings include anemia, thrombocytopenia, mild liver enzyme elevations, and increased levels of angiotensin converting enzyme. Radiologic findings of an “Erlenmeyer flask” deformity of the distal femur, fractures, and lytic lesions may also be seen. Diagnosis is confirmed by the finding of reduced glucocerebrosidase activity, usually in peripheral leukocytes, in a patient with clinical features consistent with GD. Mutation analysis can be performed and may have a role in predicting clinical course.
Treatment. The goal of medical treatment of GD is to improve symptoms and quality of life while preventing irreversible damage. GD is one of very few conditions that can be treated with enzyme replacement therapy (ERT). ERT with recombinant glucocerebrosidases is the preferred treatment of patients with type 1 and type 3 GD. It is not used for type 2 GD. ERT is very expensive (approximate cost of $US 400 000 per year for a 70-kg person) and guidelines have been developed for its use. In general, it is used for type 1 GD patients presenting as children with symptoms, or in any patient with severe disease (i.e. platelets <60 000 × 109/L, liver greater than 2.5 times normal size, spleen greater than 15 times normal size, or radiologic evidence of skeletal disease). ERT is also recommended for patients with type 3 GD who present before the onset of any neurologic symptoms. ERT has varying degrees of effectiveness in each individual. A second-line medical therapy is substrate reduction therapy. Miglustat, an inhibitor of glucosylceramide synthetase, reduces glycoplipid accumulation by decreasing the synthesis of glucocerebroside. Finally, hematopoietic stem cell transplantation can be performed and is curative for this condition.
With respect to surgical treatment, splenectomy used to be a mainstay of therapy prior to ERT. Now, it is reserved primarily for life-threatening thrombocytopenia.
Glycogen Storage Disease
Glycogen is the stored form of glucose, and inborn errors of glycogen metabolism result from virtually all of the proteins involved in its synthesis, degradation, and regulation. There are over 10 types of this disease, with five involving the liver: types 1, 3, 4, 6, and 9 (Table 22.3).
Table 22.3 Glycogen storage diseases affecting the liver
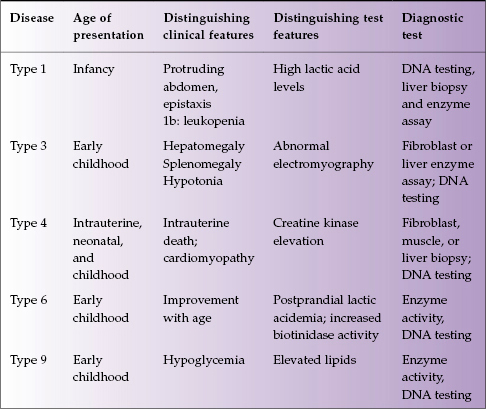
Type 1 Glycogen Storage Disease
Type 1 glycogen storage disease (GSD1) is the most common inborn error of glycogen metabolism. It results from a deficiency of a two-component enzyme system involved in the transport of glucose-6-posphate into endoplasmic reticulum and subsequent cleavage by glucose-6-phosphatase. Most patients with GSD1 present in infancy with growth failure, a protruding abdomen, hypotonia, and delayed psychomotor development. Physical examination reveals hepatomegaly, which results from increased glycogen storage in the liver. Metabolic derangements include high lactic acid levels, metabolic acidosis, low phosphorus levels, and hyperuricemia. In addition to hepatomegaly, patients demonstrate mild elevations in aminotransferases. The most accurate diagnostic measure is direct analysis of glucose-6-phosphatase enzyme activity performed on fresh liver tissue; however, histochemical staining for glucose-6-phosphatase can be highly informative. Patients with GSD1 are prone to hepatic adenomas. The risk of such adenomas undergoing malignant transformation is poorly defined but certainly not negligible.
Nutritional supplementation has become the mainstay of therapy for GSD1. For infants, formula that does not contain fructose or galactose should be used. Frequent daytime feedings with high-carbohydrate meals and continuous night-time enteral feedings (with uncooked corn starch) are required at a rate of approximately 8 mg/kg body weight per min. As solids are introduced, high-carbohydrate foods should be emphasized, but most children continue to need regular cornstarch supplementation throughout the day. Target preprandial blood glucose is higher than 4.3 mmol/L. Prevention of metabolic derangements can allow for normal psychological development.
GSD1 patients who develop multiple hepatic adenomas require evaluation for liver transplantation whereas a single adenoma may be suitable for resection. Tightening metabolic control sometimes slows the rate of growth of an adenoma. Renal insufficiency and problems relating to lipid metabolism persist after liver transplantation and may complicate management. Improved linear growth may occur.
There are two variant forms of GSD1.Glycogen storage disease 1b (GSD1b), due to a defect in the glucose-6-phosphatse transporter enzyme, displays neutropenia. In addition to management of features of GSD1, prophylaxis with antibiotics is recommended for GSD1b patients who have neutropenia and recurrent bacterial infections. Granulocyte-macrophage colony stimulation factor has also been used with success in these patients to reduce morbidities associated with severe bacterial infections.
Type 3 Glycogen Storage Disease
Type 3 glycogen storage disease (GSD3) results from a deficiency of glycogen debranching enzyme, which impairs the cleavage and transfer of dextrin from glucose molecules, subsequently restricting glucose release by phosphorylase. Since gluconeogenesis is not impacted by GSD3, the clinical course is milder than that of GSD1; however, hepatic fibrosis and splenomegaly can occur. Serum lactic acid and uric acid levels are normal. Aminotransferases may be mildly elevated. Hepatic adenomas can develop in up to 25% of these patients. Muscle weakness is often a prominent feature of this condition, which worsens with activity. The diagnosis can be confirmed by direct enzyme analysis of muscle or liver tissue of peripheral leukocytes.
Medical treatment is dietary. A high-protein and low-carbohydrate diet has been recommended to normalize metabolic activity. This diet can allow adequate gluconeogenesis and reduces the need for glycogen storage. In cases of patients with refractory hypoglycemia, night time continuous feeding of corn starch may be needed.
Type 4 Glycogen Storage Disease
Type 4 glycogen storage disease (GSD4) is a deficiency of the branching enzyme. It results in glycogen and amylopectin accumulation in hepatocytes. It is quite rare. Clinical presentation varies; a severe congenital form results in fetal hydrops, neonatal hypotonia, and fetal death, while a childhood subtype manifests as cardiomyopathy and abnormal neuromuscular development. The diagnosis of GSD4 can be made by direct enzyme analysis of liver tissue or fibroblasts. There is no known effective treatment and most patients die within the first 3 years. High-protein and low-carbohydrate diets have been used and are associated with improved growth but do not prevent liver involvement. Liver transplantation appears to be the best strategy. It has been used successfully to prolong life.
Types 6 and 9 Glycogen Storage Disease
These two types are glycogen storage disease are somewhat related. Type 6 glycogen storage disease (GSD6) is a deficiency of the liver isoform of phosphorylase. Patients typically present early in life with hepatomegaly and growth retardation. The disease improves with age and hepatomegaly resolves. Type 9 glycogen storage disease (GSD9) is a deficiency of phosphorylase kinase. Clinical manifestations are similar to that of GSD6 with hypoglycemia as a more prominent feature. Cirrhosis may occur, predominantly with GSD9.
Cystic Fibrosis
Thanks to the increased effectiveness of respiratory and nutritional treatment in childhood cystic fibrosis (CF), more children are surviving to adulthood. Chronic liver disease has become the second most common cause of death in CF. The mutant protein in CF, encoded by the cystic fibrosis transmembrane conductance regulator (CFTR) gene, is expressed in the biliary and gallbladder epithelium. CFTR is not expressed in hepatocytes and other liver cells. CFTR gene mutations affect the c-AMP inducible chloride channels in bile duct epithelium.
While the epidemiology of liver disease in CF is not well characterized, post-mortem series have demonstrated hepatobiliary pathology in up to 72% of patients. Amongst living patients, approximately 30% will have liver enzyme abnormalities, and 1–2% will develop cirrhosis. The most severe liver disease tends to occur in the pediatric population, and hence most liver transplantation for CF is in the pediatric group.
Despite all patients with CF having abnormalities in bile flow (bile tends to be thick and viscous leading to obstruction), only some develop disease. Hence, other genes may also play a role. A spectrum of hepatobiliary lesions are seen in CF. Presentation in infancy is usually as cholestatic jaundice and transient hepatomegaly. Presentation at any age can also include asymptomatic elevations in liver enzymes, hepatosplenomegaly, GI bleeding, or abdominal pain. Diagnosis can usually be made clinically or with appropriate imaging tests (ultrasound, CT, MRI/magnetic resonance cholangiopancreatography (MRCP)) and liver biopsy is rarely needed. Consequences of CF-associated liver disease include hepatic steatosis, focal biliary cirrhosis, diffuse cirrhosis, cholelithiasis and cholecystitis, secondary sclerosing cholangitis, and bile duct stenosis. Once cirrhosis develops, the usual complications can occur.
Management of CF-associated liver disease includes nutritional correction of deficiencies of fat soluble vitamins (A, D, E, K), essential fatty acids, carnitine, and choline. Ursodeoxycholic acid (UDCA) has been used as a specific therapy to delay the progression of liver disease and is administered at a dose of 20 mg/kg per day. There is no consensus on which patients will benefit the most from UDCA, and whether it should be used for early disease. However, early administration may delay the onset of liver disease.
Patients with end-stage liver disease should be evaluated for liver transplantation. The overall outcomes of well-selected CF patients post liver transplant appear to be similar to other patients.
Multidrug Resistance Protein 3 Deficiency
Although most of the recognized genetic defects of bile formation are very rare, multidrug resistance protein 3 (MDR3) deficiency (also known as progressive familial intrahepatic cholestasis type 3, PFIC3) is fairly common and has a wide range of clinical presentations, including subtle liver disease in adults. MDR3 is a P-glycoprotein thought to replenish phosphatidylcholine in the outer lipid membrane of the bile canaliculus. In the absence of MDR3 (homozygous disease), bile acids damage the canalicular membrane resulting in destruction of small bile ducts. This condition is associated with marked elevations in γ-glutamyl transpeptidase (GGT) and can lead to biliary cirrhosis. Heterozygous mutations in the ABCB4 gene coding for MDR3 can result in higher risk for cholesterol stones, sludge, and low-phospholipid associated cholestasis.
Patients with homozygous ABCB4 mutations usually present in childhood with cholestasis, jaundice, and failure to thrive. However, delayed presentation in adolescence or adults has been reported. Patients presenting with cholelithiasis prior to age 40, sludge or microlithiasis, and recurrent stones after cholecystectomy should be suspected to have heterozygous ABCB4 mutations. Many patients report a history of intrahepatic cholestasis of pregnancy, or cholelithiasis in a first-degree relative. Overall, one of every three adult patients presenting with cholestasis of unknown origin has an ABCB4 mutation.
Treatment of these patients should include nutritional correction of the fat-soluble vitamins. For patients with recurrent stones, UDCA may help prevent formation and can be used at a dose of 15 mg/kg per day. Pruritus can be severe and often unresponsive to conventional therapies such as UDCA, cholestyramine, antihistamines, and rifampin. In refractory cases, surgical interruption of the enterohepatic circulation may be necessary. Liver transplantation is reserved for patients with end-stage liver disease or refractory pruritus.
Hereditary Citrullinemia Type 2
Unlike citrullinemia type 1 (a urea cycle disorder discussed below), type 2 citrullinemia occurs in both adults and children. Type 2 citrullinemia has a prevalence of 1 in 100 000 to 1 in 230 000 individuals. In infants, it may cause a neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). In adults, it can resemble non-alcoholic fatty liver disease clinically, though the primary organ targeted is the nervous system (symptoms include confusion, lethargy, hallucinations, delusions, tremors, and coma). Treatment of choice is liver transplantation for advanced disease.
Urea Cycle Defects
The urea cycle consists of five enzymes that process ammonia derived from amino acid metabolism to urea. Genetic defects have been reported in all of these enzymes. The syndromes caused by these defects are not associated with severe liver disease, but they can mimic other metabolic liver diseases. All of the urea cycle disorders present as acute, life-threatening events in infancy and early childhood, though adult presentations have also been reported. A common early sign in newborns is central hyperventilation leading to respiratory alkalosis. Clinical manifestations vary significantly (adults may present only with neurologic symptoms), but all affected have very high serum venous and arterial ammonia levels (may exceed 2000 mmol/L, normal <50 mmol/L). Tests performed should include serum ammonia, arterial blood gases (expect a respiratory alkalosis), urine organic acids, serum amino acids, and urinary orotic acid. Direct enzyme analysis in liver tissue, fibroblasts, and red blood cells can be performed and is useful in patients presenting in adulthood.
Treatment of urea cycle defects is mainly medical. The immediate goal of therapy in patients presenting with newly diagnosed urea cycle defects is to restore ammonia levels in blood to normal. Oral lactulose can be used but may not be effective given the high levels of serum ammonia. Usually hemodialysis or hemofiltration is required. Alternative metabolic pathways for waste nitrogen disposal can be utilized by intravenous administration of sodium benzoate (caution in cirrhotic patients in whom a paradoxical rise in serum ammonia can be seen) and sodium phenylacetate. Once a patient stabilizes, long-term therapy with protein restriction can be initiated and individually tailored. Essential amino acid supplementation is necessary. Liver transplantation is recommended for patients who deteriorate despite therapy. Transplanted patients have normalization of their enzyme activity and ammonia levels and can tolerate a normal diet. Some patients with severe enzyme deficiencies may benefit from early transplantation, even though the liver is structurally normal.
Muscular Dystrophies
Duchenne muscular dystrophy and other genetic disorders classified as muscular dystrophy do not affect the liver. Some patients, mainly young children, present clinically with extremely subtle muscular involvement but prominently abnormal serum aminotransferases, usually AST greater than ALT. When it is established that the liver is normal, finding an extremely elevated serum creatine phosphokinase (CPK) can point to the correct diagnosis. Consultation with a neurologist or specialist in muscle disease is the next step in clinical management.
Metabolic Liver Disorders Presenting As Conjugated Hyperbilirubinemia in Early Infancy
At least one-quarter of all infants who develop conjugated hyperbilirubinemia in the first 3 months of life have a metabolic liver disease. A few hepatic metabolic disorders, like the urea cycle defects, may present without conjugated hyperbilirubinemia. Any infant presenting with neuromuscular disease, developmental delay, failure to thrive, hepatomegaly, or elevated serum aminotransferases should be evaluated for acidosis, hypoglycemia, bleeding diathesis, and hyperammonemia. Parental consanguinity, recurrent miscarriages, or early infant deaths of previous siblings suggest inherited liver disease. A family pedigree displaying unexplained liver disease, neurologic disease, or muscle disease should also raise suspicion for inherited metabolic problem.
Not all hepatic metabolic diseases present with conjugated hyperbilirubinemia in the neonatal period. There is an extensive differential diagnosis of non-metabolic disorders that also can present with conjugated hyperbilirubinemia at this time, such as biliary atresia, certain congenital infections, and Alagille syndrome. Therefore methodical and extensive clinical evaluation is important. Early, accurate diagnosis is always important for best clinical outcome.
Selected diseases of note are described in Table 22.4.
Table 22.4 Selected metabolic liver diseases presenting in infancy with conjugated hyperbilirubinemia
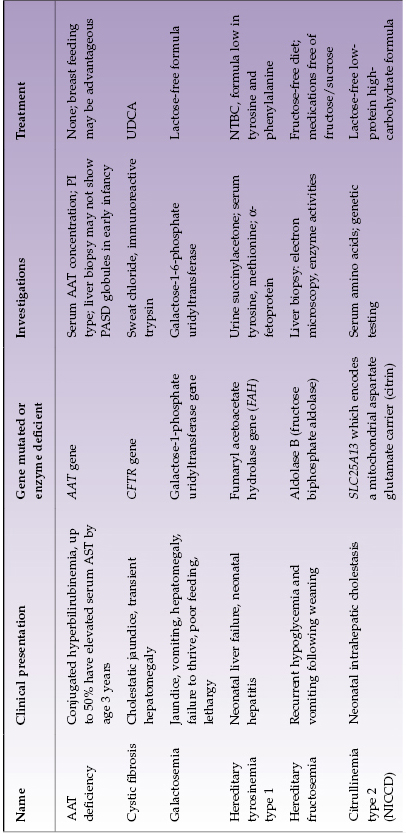
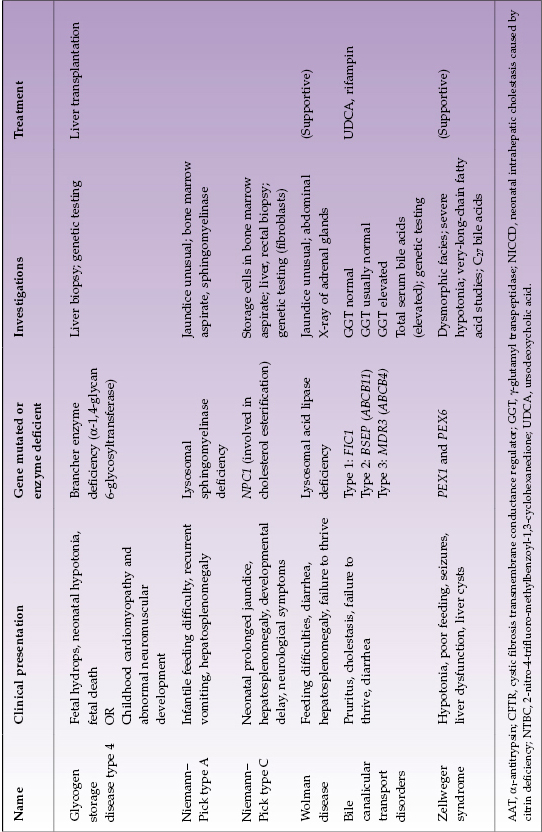
With disorders that can present in infancy or later, affected infants often have some distinctive features. In AAT deficiency approximately one-quarter of infants have very severe cholestasis. They may have acholic stools and, exceptionally, infants are found to have both AAT deficiency and biliary atresia. Infants whose jaundice is severe and slow to resolve generally have a poor overall prognosis and need early liver transplantation. Likewise, some infants with citrulinemia type 2 (citrin deficiency) have very severe neonatal hepatitis and have some metabolic features suggestive of galactosemia. Differentiation from galactosemia is important because treatment of the two disorders differs. Infants with CF may present with severe neonatal hepatitis with ductopenia or extrahepatic bile duct damage approximating biliary atresia or severe fatty liver. Those with meconium ileus are more likely to have liver involvement. Of the glycogen storage disorders, type 4 is the only one that presents with neonatal conjugated hyperbilirubinemia.
Galactosemia
Galactose is a sugar found in human and bovine milk primarily as the disaccharide lactose. Altered metabolism of galactose caused by deficient enzyme activity results in elevations in blood levels of galactose, called galactosemia. Galactosemia can result from three different enzyme deficiencies: galatose-1-phosphate uridyl transferase (GALT), galactokinase (GALK), and uridine diphosphate galactose-4-epimerase (GALE). Each enzyme deficiency has a different phenotype.
Classic galactosemia is caused by severe deficiency of GALT. It is the most common and severe. Affected individuals usually present in the first few days of life with jaundice, vomiting, hepatomegaly, failure to thrive, poor feeding, and lethargy. Examination can reveal hypotonia and lenticular cataracts. Laboratory abnormalities will include increased plasma galactose, increased blood and urine galactitol levels, liver dysfunction, metabolic acidosis, and hemolytic anemia. Gold standard for diagnosis is demonstration of absence of GALT activity in red blood cells.
GALK and GALE deficiency are far less common. GALK deficiency presents with lenticular cataracts and rarely as pseudotumor cerebri. Deficiency of GALE is restricted to erythrocytes and so systemic disease is not seen.
Management of galactosemia includes minimal intake of dietary galactose (by excluding milk and dairy products from the diet), use of soy-based infant formula, and calcium supplementation. If identified early (by a newborn screening program) and adequate dietary therapy is instituted immediately, most patients will have a normal childhood. However, there still remains an increased risk of neuropsychological and ovarian problems in teenage years.
Hereditary Tyrosinemia Type 1
Hereditary tyrosinemia type 1 (HT1) is an autosomal recessive condition caused by an enzymatic defect in fumarylacetate hydrolase, which leads to accumulation of the intermediates fumarylacetoacetate and maleylacetoacetate, which are then converted to toxic metabolites within the liver. Patients usually present with neonatal liver failure or chronic liver disease. Some infants, however, present with neonatal hepatitis and a few present with unexplained chronic liver disease with hepatic synthetic dysfunction later in the first 6 months of life. Typical biochemical features include abnormal standard tests of liver function, elevated levels of tyrosine and methionine, and serum α-fetoprotein is greatly elevated in the 40 000–70 000 IU/L range. The most informative test is the presence of succinylacetone in the urine. Those with the chronic form of HT1 have milder features and usually present after age 1 year old. These children may have hepatomegaly, rickets, nephromegaly, and systemic hypertension. Acute neuropathy resembling a porphyric crisis may occur. Hepatocellular carcinoma may develop in any child with HT1. In addition to the amino acid profile and urinary succinylacetone, definitive diagnosis of this condition is confirmed by genetic testing or measuring fumarlyacetate hydrolase levels in lymphocytes, erythrocytes, or liver tissue.
Treatment of tyrosinemia has been revolutionized by the introduction of NTBC (2-nitro-4-trifluoro-methylbenzoyl-1,3-cyclohexanedione) in the 1990s. NTBC, a herbicide, is a potent inhibitor of one of the initial steps in the degradation of tyrosine. The downstream toxic intermediates therefore do not accumulate. Nutritional therapy is still required with NTBC treatment. It consists of removing tyrosine and phenylalanine from the diet. This dietary restriction minimizes adverse effects of NTBC treatment, prevents or reverses renal damage, and improves metabolic bone disease.
Liver transplantation corrects the phenotype of this disease and restores normal fumarylacetate hydrolase activity. Previously, liver transplantation was one of the mainstays of management but this need has greatly declined with the use of NTBC.
In places where HT1 is highly prevalent, newborn screening programs have proved highly effective. Infants are diagnosed very early in life and started on NTBC. Early treatment with NTBC has been very successful at improving the hepatic clinical course of patients with tyrosinemia. With delayed treatment, however, the risk of hepatocellular carcinoma persists.
Hereditary Fructose Intolerance (Fructosemia)
Fructosemia is an autosomal recessive disorder due to the deficiency of fructose-1-phosphate aldolase, isozyme b. Deficiency of this enzyme leads to the accumulation of fructose-1-phosphate and subsequent inhibition of hepatic phosphorylase and gluconeogenesis. As a result, patients present with recurrent hypoglycemia and vomiting following weaning, which is when fructose or sucrose is typically added to an infant’s diet. Some infants who are formula fed may present earlier due to the sucrose content of some brands. As disease progresses, additional manifestations include jaundice, hepatomegaly, lactic acidosis, fructosuria, aminoaciduria, and failure to thrive.
Diagnosis has historically required a liver biopsy and measurement of enzyme levels. However, biopsy can now generally be avoided due to the availability of genetic testing of the aldolase B gene on chromosome 9q. Therapy involves elimination of fructose from the diet, which can be difficult since it involves eliminating sucrose. Pharmacists can advise as to medications free of sucrose. Adherence to this diet will tend to reverse and prevent additional manifestations of disease.
Neonatal Liver Failure
Some metabolic disease cause severe liver disease which presents as neonatal liver failure (Table 22.5). The clinical picture may resemble adult acute liver failure or end-stage chronic liver failure, the latter apparently due to liver disease beginning antenatally in the fetus. Early recognition and intervention are critically important for such infants. Management should include elemental formula to remove potentially toxic sugars from the diet; exclusion of tyrosine and phenylalanine may also be instituted pre-emptively. Nutritional therapy can be revised once the relevant diagnoses have been ruled out. Since both bacterial and viral infection can cause a similar picture, diagnosis and management of infection are important. Perinatal hemochromatosis, which is usually due to an alloimmune process, can be investigated by serum ferritin, MRI of liver and abdominal organs, and bone marrow inspection to exclude hemophagocytosis. Perinatal hemochromatosis usually, but not invariably, affects the second and subsequent children in a family.
Table 22.5 Metabolic disorders that can present as neonatal liver failure resembling acute liver failure (“acute pattern”) or chronic liver failure (“chronic pattern”)
| Acute pattern (Jaundice, extremely elevated serum aminotransferases, fixed coagulopathy) | Chronic pattern (Jaundice, near-normal serum aminotransferases, low serum albumin, fixed coagulopathy) |
| Hereditary tyrosinemia, type 1 | Hereditary tyrosinemia, type 1 |
| Galactosemia | Galactosemia Hereditary fructosemia |
| Mitochondrial disorders | Mitochondrial disorders Inborn error of bile acid synthesis: Δ4-3-oxosteroid-5β-reductase Perinatal hemochromatosis |
Related posts:
 9 Indications for liver transplantation in adults and children
9 Indications for liver transplantation in adults and children
 16 Drug-induced liver disease
24 Liver disease in pregnant women
16 Drug-induced liver disease
24 Liver disease in pregnant women
 14 Chronic viral hepatitis in adults and children: hepatitis C
14 Chronic viral hepatitis in adults and children: hepatitis C
 12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
 26 An internist’s approach to radiologic examination of the liver
26 An internist’s approach to radiologic examination of the liver



