- Portal vein thrombosis is common in patients with cirrhosis.
- The main complication of portal vein thrombosis is variceal bleeding.
- Acute portal vein thrombosis without cirrhosis should be treated with anticoagulation.
- Anticoagulation in the setting of cirrhosis for portal vein thrombosis remains controversial but is likely safe.
- Budd–Chiari syndrome is almost always associated with an underlying predisposing condition (JAK2 tyrosine kinase mutations are the most common).
- Acute Budd–Chiari syndrome is a medical emergency.
- Transjugular intrahepatic portosystemic shunt (TIPS) is first-line therapy for acute Budd–Chiari syndrome.
- Sinusoidal obstruction syndrome (formerly called veno-occlusive disease) is usually a complication of stem-cell transplantation.
- Treatment options for sinusoidal obstruction syndrome are limited but prophylactic ursodeoxycholic acid appears beneficial.
Vascular Anatomy
The liver is unique in that its primary blood supply is venous blood entering the liver through the portal vein. The hepatic artery supplies 30% of the blood volume but provides 90% of the oxygenated blood to the liver. The veins of the entire gastrointestinal tract drain into the portal vein, which then branches into the left and right portal vein as it enters the liver. The portal vein branches into smaller and smaller units until portal venules, which supply individual portal tracts, finally flow into the hepatic sinusoids.
The sinusoids are lined by sinusoidal epithelial cells but are not true blood vessels as the sinusoids are fenestrated, allowing nutrients and waste to diffuse between the blood stream and the hepatocytes. The sinusoids drain into hepatic venules, which drain into larger and larger hepatic veins, which drain into the right, middle, and left hepatic veins.
The hepatic veins drain into the inferior vena cava (IVC). The caudate lobe of the liver has its own venous outflow, which empties directly into the IVC. As a result, in the setting of hepatic vein outflow obstruction (e.g. Budd–Chiari syndrome), the caudate lobe enlarges as blood from the other lobes of the liver is rerouted to the caudate to allow it to drain into the IVC and thereby return to the heart.
Portal Vein Thrombosis—with Cirrhosis
Cirrhosis is present in 25–28% of cases of portal vein thrombosis (PVT). PVT is common in patients with cirrhosis, particularly as the disease progresses. Sluggish flow through the liver and possibly an imbalance between prothrombotic and antithrombotic factors in the liver increase the risk of PVT in patients with cirrhosis.
Although typically patients with cirrhosis are thought to be at increased risk of bleeding, it is important to note that although the liver makes clotting factors, it also makes antithrombotic factors such as protein C and protein S. Even when the PT/INR is elevated (this is the only parameter that is easily measurable), patients may actually be hypercoagulable.
Thrombophilic tendencies are identified in 40 to 70% of patients with cirrhosis and PVT. These include the Factor V Leiden mutation as well as mutations in the methylene–tetrahydrofolate and prothrombin genes.
Prevalence data estimate 10–25% of patients with cirrhosis have PVT.1 Old studies suggest an incidence of less than 1% per year in patients with compensated cirrhosis, but PVT occurs much more frequently with more advanced liver disease. In patients on the liver transplant waiting list, PVT develops in 7 to 16% annually. The risk of PVT is highest with hepatocellular carcinoma (HCC), with rates as high as 35%. In contrast, PVT occurs in 16% of alcohol or viral-related cirrhosis, 8% in primary biliary cirrhosis, and 3.5% in primary sclerosing cholangitis.2
Presentation
Acute
Acute PVT is very variable, ranging from an asymptomatic incidental finding on imaging to precipitation of acute life-threatening hepatic decompensation. Symptoms may include GI bleeding from varices or portal hypertensive gastropathy (approximately 40%) and abdominal pain (approximately 18%) with possible extension to the mesenteric veins with intestinal infarction (a potentially fatal complication). Ascites is relatively uncommon unless patients already have advanced cirrhosis. Ascites may develop with fluid resuscitation after a GI bleed.
It may be difficult to differentiate whether symptoms are from PVT or progression of underlying advanced cirrhosis.
Chronic
Typically, chronic PVT is found incidentally on radiologic investigation done for another purpose (e.g. HCC surveillance); however, it may also lead to a symptomatic presentation, usually with acute variceal hemorrhage. Esophageal varices are present in 85–90% of patients with PVT and 30–40% also have gastric varices. Variceal hemorrhage occurs at least once in 50–70% of patients over time and in 30% of patients on more than one occasion. Varices may also develop in atypical locations. Isolated splenic vein thrombosis usually leads to the development of just gastric varices. Splenomegaly is common with PVT and may be massive.
Diagnosis
The diagnosis is usually made with ultrasonography with Doppler evaluation. Contrast-enhanced ultrasonography (if available) is more sensitive but unnecessary in most cases.
Computed tomography (CT) or magnetic resonance imaging (MRI) allow better evaluation of the extent of the PVT and also may be used to evaluate for the presence of HCC in the liver and/or portal vein. Spontaneous shunts may develop between the splenic vein and left renal vein to decompress the portal system, which also are better visualized with CT or MRI.
Prognosis
The natural history of PVT in cirrhosis is not well documented. In a study of 37 patients with non-malignant PVT in the setting of cirrhosis, 5-year survival was only 45%.3
Recanalization of both acute and chronic PVT may occur. If recanalization occurs with acute PVT, there may be no evidence of its past occurrence and recurrence appears to be relatively infrequent. In the presence of cirrhosis, recanalization of the portal vein is uncommon. Cavernous transformation, in which collateral vessels develop to maintain blood flow in a hepatopedal direction, is typical of non-cirrhotic PVT. In contrast, with cirrhosis, PVT may persist or alternatively a spontaneous shunt may develop between the portal vein and the left renal vein, which decompresses the portal system but may lead to severe encephalopathy, out of keeping with the degree of liver dysfunction, due to marked portosystemic shunting.
PVT may progress from the large vessels to the small intrahepatic veins. Even if recanalization of the large vessels subsequently occurs, the intrahepatic microthrombi may lead to worsening of cirrhosis due to so-called “parenchymal extinction”.
A trial of prophylactic low-molecular weight heparin (LMWH) (doses similar to deep vein thrombosis prophylaxis) in patients with advanced cirrhosis to prevent PVT showed that not only did patients have a lower incidence of PVT but also a lower rate of hepatic decompensation, reinforcing the concept that microthrombi in the liver may worsen cirrhosis.4 Whether patients awaiting liver transplant should receive daily prophylactic heparin is currently unknown.
Management
Optimal management of PVT in the setting of cirrhosis is unknown. No randomized controlled trials have been performed. Anticoagulation is the most common treatment employed, with the primary goal of preventing extension of the clot rather than necessarily achieving recanalization (Fig. 21.1). In acute PVT without cirrhosis, early introduction of anticoagulation is advised, with 80% recanalization and a reduced risk of future thrombotic events or progression of clot to the mesenteric system. In patients with cirrhosis, the main concern is that if variceal hemorrhage were to occur, bleeding may be much more severe with anticoagulation. However, anticoagulation is unlikely to directly precipitate variceal hemorrhage. In a small study of 29 patients on the liver transplant waiting list, anticoagulation led to PVT recanalization in 8 of 19 compared to 0 of 10 who were left untreated. Transplant outcome was also improved, likely because the presence of PVT significantly complicates liver transplant surgery.5 Anticoagulation with LMWH may be preferred to warfarin. Warfarin affects the INR and therefore makes the MELD score unreliable. Although a modification of MELD is made for patients on anticoagulation, LMWH may have other advantages such as less need for monitoring because of weight-based dosing. New oral anticoagulants may offer similar advantages; however, their effects cannot be easily reversed with fresh frozen plasma and therefore should be used with caution in patients with cirrhosis.
Fig. 21.1 Management of acute portal vein thrombosis (PVT). *Optimal anticoagulant is unclear—consider warfarin, low-molecular weight heparin, or new oral anticoagulant agents.
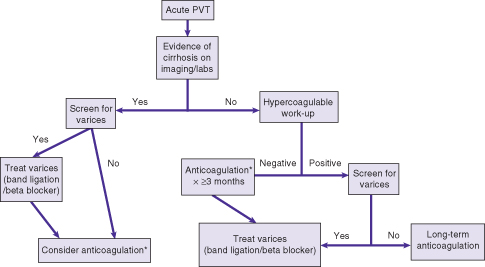
If anticoagulation is used, variceal screening should be performed. The optimal therapy for varices in the setting of PVT with anticoagulation is unknown (beta-blockers vs. band ligation). Some advocate band ligation prior to starting anticoagulation, particularly if the varices have high-risk features (e.g. red spots). The risk of such an approach is bleeding from postbanding ulceration. Other therapeutic options for PVT include thrombectomy (minimal data) and transjugular intrahepatic portosystemic shunt (TIPS). Thrombectomy is followed by intraportal thrombolysis. TIPS is feasible if the intrahepatic PV branches are patent. TIPS placement is followed by anticoagulation and should serve as a bridge to liver transplantation.
Portal Vein Thrombosis—without Cirrhosis
Approximately 75% of patients with PVT do not have underlying cirrhosis. The most common causes of non-cirrhotic PVT are abdominal surgery, abdominal sepsis (usually in childhood), pancreatitis, and hypercoagulability. Myeloproliferative disorders are the most common thrombophilic states seen with PVT. PVT may also occur due to childhood infections such as omphalitis. Neoplastic causes of PVT with tumor thrombus, particularly from renal cell carcinoma, should be excluded with imaging.
Presentation
Like in cirrhosis, PVT ranges from asymptomatic to life-threatening if intestinal infarction occurs from clot extension. Development of varices is common, with a high risk of bleeding. Variceal hemorrhage is much better tolerated in patients with non-cirrhotic PVT because of preserved liver synthetic function. In addition to atypical varices, patients often develop splenomegaly, which may be massive. Cavernous transformation, in which collateral vessels develop to maintain blood flow in a hepatopedal direction, often develops, particularly when PVT occurs in childhood. Cavernous transformation is usually easily seen on abdominal imaging. Even with recanalization or cavernous transformation, the spleen may remain enlarged. If so, patients, particularly children and adolescents, should be advised to avoid contact sports or be fitted with a splenic guard to prevent splenic rupture.
Prognosis
The major problem with non-cirrhotic PVT is variceal bleeding. Long-term survival is excellent with a 5-year mortality rate of 5 to 10%. Complications and fatalities are more commonly due to the underlying illness precipitating the PVT than to complications of the PVT itself.
In acute non-cirrhotic PVT, anticoagulation is recommended. Therapy should be continued for at least 3 months, but if a hypercoagulable risk factor is identified, longer-term (potentially indefinite) anticoagulation is necessary. Recanalization is reported to occur in from 40% to as high as 80% of patients when treatment is started at diagnosis.6
Anticoagulation is likely less useful in chronic PVT. The largest study showed a higher risk of bleeding (12.5 per 100 patient years) than thrombosis (5.5 per 100 patient years) in patients anticoagulated for chronic non-cirrhotic PVT.7 Guidelines recommend anticoagulation for those with non-cirrhotic PVT and an identifiable thrombophilic predisposition that cannot be corrected. Screening for varices with institution of appropriate therapy (beta-blockers or band ligation) is recommended prior to starting anticoagulants. Band ligation is usually preferable as there are no data on beta-blocker therapy in PVT and, as the pathophysiology is different from that for varices secondary to portal hypertension due to cirrhosis, the efficacy may differ.
Portal Biliopathy
Patients with chronic PVT (usually without cirrhosis) may develop bile duct abnormalities visible on cholangiogram that typically present with cholestatic jaundice. Biliary findings on imaging include: biliary stenosis, upstream dilatation, wavy appearance to the biliary tree, and angulation of the common bile duct. Gallbladder and choledocal varices are common in patients with biliopathy, however bleeding is very uncommon.
Portal biliopathy is thought to occur in the setting of long-standing PVT, but the precise mechanisms responsible are not well understood. Ursodeoxycholic acid is used empirically to treat portal biliopathy. Endoscopic retrograde cholangiopancreatography (ERCP) with stenting may be useful if significant strictures develop. Rarely mesocaval or other surgical shunts are performed to increase portosystemic shunting to reduce portal inflow and thus portal pressure.
Budd–Chiari Syndrome
Budd–Chiari syndrome (BCS) usually refers to a thrombosis of the hepatic veins or suprahepatic inferior vena cava (see also Chapter 24). The largest series of BCS included 237 patients from four centers in three countries. It was found that BCS was more common in woman (67%), especially during or shortly after pregnancy, and the median age of presentation was 35 years old (range 13–76). Obstruction was in the hepatic veins in 62%, the IVC in 7%, and both in 31%, and 14% had associated PVT.8
Presentation
Patients may present with acute, subacute, or chronic BCS.
- Acute (20%): usually very symptomatic, classically with new-onset ascites, severe right upper quadrant pain, and tender hepatomegaly that may progress to fulminant hepatic failure. Mixed elevation of alanine aminotransferase (ALT) and alkaline phosphatase (ALP), with subsequent development of jaundice, is common.
- Subacute (40%): symptoms for less than 6 months with no evidence of cirrhosis.
- Chronic (40%): signs and symptoms for more than 6 months with evidence of portal hypertension and cirrhosis at diagnosis.
Subacute and chronic BCS are often only mildly symptomatic or entirely asymptomatic. Some patients recall a period of abdominal pain with or without distension in the past. The chronic congestion associated with hepatic outflow obstruction leads to cirrhosis. In chronic BCS, the caudate lobe hypertrophies because of shunting of blood from the rest of the liver to the caudate lobe to allow drainage through its unique direct connection to the IVC. With time, the hypertrophied caudate lobe may compress the IVC, worsening the venous return and increasing portal hypertension.
Complications of cirrhosis may develop and notably up to 28% of patients have been reported to develop the “hepatopulmonary syndrome” with intrathoracic right to left shunts. The explanation for the association between BCS and hepatopulmonary syndrome is unknown.
Etiology
An underlying disorder is identified in more than 80% of patients (Box 21.1). Up to half may have more than one thrombophilic abnormality. The most common conditions associated with BCS are the myeloproliferative disorders, with polycythemia vera occurring most frequently.9
Related posts:
 9 Indications for liver transplantation in adults and children
9 Indications for liver transplantation in adults and children
 16 Drug-induced liver disease
24 Liver disease in pregnant women
16 Drug-induced liver disease
24 Liver disease in pregnant women
 14 Chronic viral hepatitis in adults and children: hepatitis C
14 Chronic viral hepatitis in adults and children: hepatitis C
 12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
12 Acute viral hepatitis in adults and children: hepatitis A, B, C, D, E, and others
 26 An internist’s approach to radiologic examination of the liver
26 An internist’s approach to radiologic examination of the liver



