John C. Pope, IV, MD
Many of these conditions are related to maldevelopment of the kidney and urinary tract and are thus the result of anomalous genetic mechanisms, some of which are understood, others of which are not. A basic knowledge of molecular genetics is required to better understand normal renal development, absence of renal development, and the effects of genetic mutations and abnormal signaling proteins on renal maldevelopment and cystic diseases. Within this chapter, the relevant genetic processes will be discussed as they relate to specific disease processes. Further discussion on the specific genes involved with renal development appears in Chapter 111 and in an article by Glassberg in 2002.
Overview of Genetics
Each gene within a cell is one of a set of two; each is called an allele. One defective allele may be the carrier for the defect. In Knudson’s (1991) two-hit theory, the first hit is an inherited mutation that is found in all cells of an individual (i.e., a germline mutation). The second hit occurs when the wild-type, normal allele spontaneously mutates within a specific organ, although it may be a different mutation than in the primarily affected allele (Knudson, 1991). For example, the typical somatic cells in the kidney and all other cells of a patient with von Hippel-Liudau (VHL) disease typically are heterozygous for the VHL gene located on chromosome 3; that is, the mutant VHL allele is inherited and its mate is the normal wild-type allele. However, the configuration of wild-type VHL makes it prone to spontaneous mutation. If the wild-type allele develops a defect within the cells of a specific organ, those cells no longer are able to produce normal pVHL (VHL protein) with its suppressor properties. That organ then has the propensity to develop a tumor. The variability in manifestations of autosomal dominant polycystic kidney disease (ADPKD) may be related to a two-hit phenomenon as well. For example, ADPKD is a heterozygous condition in which one allele of the PKD1 or PKD2 set is responsible for the genetic transmission of the disease but may not be enough for cystogenesis. A spontaneous mutation of the wild-type allele may be responsible for cystogenesis or for other manifestations of the disease. Qian and colleagues (1996) suggested that the PKD1 gene has an unusual genomic structure that makes it readily mutable for a second hit, and the timing of this second hit may account for the phenotypic variability within one family. The second hit itself may occur only in a few cells, possibly accounting for the fact that less than 1% of nephrons in ADPKD eventually develop cysts (Qian et al, 1996). Within a cyst, many but not all of the lining cells have lost their heterozygosity (Brazier and Henske, 1997; Qian et al, 1999).
Renal Agenesis and Dysplasia
There are many terms that can be used to describe anomalous development of the kidney. These terms are often used interchangeably and can result in great confusion. The definitions as adopted in 1987 by the American Academy of Pediatrics (AAP) Section on Urology (Glassberg et al, 1987) will be employed in this chapter.
Renal Agenesis
Renal agenesis, or absent kidney development, can result from anomalous development of the wolffian duct, ureteric bud, and/or metanephric blastema. Bilateral agenesis occurs in 1 of every 4000 births and has a male predominance (Potter, 1965). Fetal demise occurs secondary to the lack of fetal urine production and subsequent oligohydramnios. Affected infants are born with immature lungs and pneumothorax, Potter facies (hypertelorism, prominent inner canthal folds, and recessive chin), and orthopedic defects secondary to intrauterine compression. Most cases of bilateral agenesis are sporadic, and many are associated with other congenital anomalies, including urogenital sinus defects.
Unilateral agenesis is more common, with an incidence of 1 in 450 to 1000 births (Kass and Bloom, 1992). Again, a wolffian duct abnormality is often the cause, and associated other wolffian and müllerian abnormalities may also be present, such as malformation of the ipsilateral uterine horn, fallopian tube or ovary in the female, or absence of the ipsilateral testis, vas deferens, or seminiferous tubules in the male. Mayer-Rokitansky-Küster-Hauser syndrome refers to a group of associated findings that include unilateral renal agenesis or renal ectopia, ipsilateral müllerian defects, and vaginal agenesis. Of men with unilateral or bilateral congenital absence of the vas deferens, 26% or 11%, respectively, had renal agenesis. Congenital bilateral agenesis of the vas deferens is also an expected finding in male patients with cystic fibrosis. Cystic fibrosis transmembrane-conductance regulator (CFTR) gene mutations frequently contribute to maldevelopment of the vas deferens, but vasal agenesis can occur without any evidence of CFTR defects. CFTR abnormalities are rarely detected in men with congenital absence of the vas deferens and renal anomalies (Schlegel et al, 1996).
Unilateral renal agenesis can also be associated with other urologic abnormalities in 48% of patients, including primary vesicoureteral reflux (28%), obstructive megaureter (11%), and ureteropelvic junction obstruction (3%) (Cascio et al, 1999). These findings are similar to those associated with unilateral multicystic kidney disease, suggesting the possibility that some absent kidneys represent involuted multicystic dysplastic kidneys.
Additional reading on renal agenesis may be found in Chapter 117.
Dysplasia
Definition
Renal dysgenesis is defined as maldevelopment of the kidney that affects its size, shape, or structure. It is composed of two primary subtypes, the first of which is dysplasia. The term dysplasia, while literally and most simply is defined as “abnormal tissue,” specifically, as it relates to the kidney, it can only truly be defined on the basis of histopathology. Dysplasia is a histologic diagnosis made by the presence of embryonic, immature mesenchyme, and primitive renal components. The differentiation of the metanephros into mature renal components halts at some point along the developmental pathway, often due to an error in the wolffian duct/ureteric bud/metanephric blastema interaction. Common histologic features include distortion of renal architecture, immature or primitive glomeruli, nephron precursors such as comma and S-shaped bodies, and cartilage and tubules encircled by collars of fibromuscular cells (referred to as primitive ducts) (Fig. 118–1). Ericsson and Ivemark (1958) considered these primitive ducts to be the sine qua non finding of all dysplasia. Cysts may or may not be present. The etiology for the development of dysplasia appears to be twofold: (1) a primary, inherent abnormality in the differentiation of the nephron and collecting duct, often with an underlying genetic cause, and (2) a secondary cause resulting from congenital urinary tract obstruction.

Etiology
Renal dysplasia is the primary cause of childhood end-stage renal disease and two main theories have been considered in its pathogenesis: (1) a primary failure of ureteric bud activity and (2) a disruption in renal development produced by fetal urinary outflow impairment (obstruction). Mackie and Stephens (1975) described the ureteric bud theory suggesting that ectopic (cephalad or caudal) ureteric bud formation leads to abnormal ureteric orifice location either laterally (often leading to vesicoureteral reflux) or distally (often leading to ureteral obstruction). Normal nephrogenesis is dependent upon the ureteric bud meeting the center of the metanephric mesenchyme so that nephrogenesis is appropriately induced by the ureteric bud. However, an abnormally located ureteric bud would be expected to penetrate into a peripheral, degenerating area of the metanephric blastema. The result is a marginal (i.e., hypoplastic or dysplastic) kidney. In cases of dysplasia that are associated with an ectopic ureter, it is not clear if it is the obstruction, aberrant ureteric bud/metanephros interaction, or some other factor that leads to the dysplasia. Dysplasia associated with vesicoureteral reflux would also be included in this category.
Currently, there is no direct experimental evidence to prove that an abnormal ureteric bud site leads to ureteric bud penetration at a peripheral area of the blastema. However, there is much data to support the idea that defects in genetic and apoptotic pathways can affect ureteric bud formation, branching morphogenesis within the metanephric blastema, and normal nephrogenesis (e.g., RET, RAR, BMP4, SLIT2-ROBO2, COX-2, AT2) (Mendelsohn et al, 1994; Norwood et al, 2000; Pope et al, 2001; Grieshammer et al, 2004; Thomas et al, 2005). The genetic defect at each point may be the same, but the downstream pathway associated with the defect may vary for each process.
Animal studies of fetal urinary tract obstruction generate some but not all of the anatomic and histopathologic features of human renal dysplasia. Findings in these animal studies include dilatation at various segments of the nephron and collecting duct, inhibition of nephron development in the nephrogenic zone, architectural disorganization, and the appearance of primitive glomeruli as well as S-shaped bodies and cysts. At times, obstruction in these animal models can even lead to dedifferentiation of certain cells. An example is the conversion of renal epithelial cells back into mesenchymal cells and even of mesenchymal cells into myofibroblasts (Peters et al, 1992; Nguyen et al, 1999; Matsell and Tarantal, 2002). Dysplasia arising from obstruction typically appears at the periphery of the kidney, at the nephrogenic zone, and frequently with subcapsular cysts. This type usually develops later in gestation, such as in the case of posterior urethral valves (PUVs) (Fig. 118–2).

Ultrasonographically, dysplastic kidneys have a distinct appearance. The kidney is typically small and is hyperechogenic when compared with the liver. There is loss of the typical corticomedullary differentiation, and there is minimal distinction of the renal parenchyma and the perirenal fat (Fig. 118–3A). Cysts can be seen in conjunction with parenchymal dysplasia (Fig. 118–3B). These cysts can also develop from either abnormal development of the metanephric blastema or from distal obstruction. Again, the exact etiology of the cyst formation can be unclear. The important point is that not all cysts are a result of dysplasia and not all dysplasia is associated with cysts.
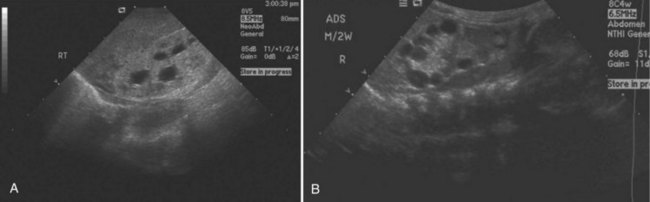
(Courtesy of Marta Hernanz-Schulman, MD.)
Hypoplasia and Hypodysplasia
Renal Hypoplasia
Hypodysplastic kidneys most often occur in conjunction with ectopic ureteric orifices, with the extent of dysplasia correlating with the degree of ectopia (Schwarz et al, 1981). However, hypodysplastic kidneys are seen in a few patients with normal ureteric orifices. In such cases, obstruction may or may not be present.
Oligomeganephronia
First described in 1962 (Habib et al, 1962; Royer et al, 1962), oligomeganephronia is a type of renal hypoplasia that results from a quantitative defect of the renal parenchyma with a reduced number of nephrons and hypertrophy of each nephron. This condition differs histopathologically from simple hypoplasia in which the renal mass is reduced but the number of nephrons is normal. Oligomeganephronia may occur as a sporadic defect or in association with a number of syndromes. It is usually a bilateral condition, although a few instances of unilateral oligomeganephronia associated with contralateral renal agenesis have been reported (Griffel et al, 1972; Lam et al, 1982; Forster and Hawkins, 1994).
Etiology
Oligomeganephronia results from arrested development of the metanephric blastema at 14 to 20 weeks’ gestation, with subsequent hypertrophy of glomeruli and tubules in the kidney. The hypertrophy and hyperfiltration results in further nephron injury and sclerosis. Eventually, this progressive loss of nephrons leads to end-stage renal disease (ESRD). Although oligomeganephronia is associated with some genetic syndromes, most cases are sporadic. However, mutations in the paired-box transcription factor (PAX2), have been noted in persons with nonsyndromic oligomeganephronia (Salomon et al, 2001). Recently, mutations in the homeobox transcription factor (hepatocyte nuclear factor-1b) have been described in association with oligomeganephronia (Bohn et al, 2003). Interestingly, the heterozygous mutation may be associated with development of the kidney lesion. Vascular abnormalities and accidents have been associated with this type of renal hypoplasia. The cause of most oligomeganephronia cases is unknown.
Histopathology
Meticulous histologic examination of the kidney is the only way to establish an absolute diagnosis of oligomeganephronia. There is a reduced number of nephrons, which are all elongated (sometimes fourfold) and widened, particularly at the proximal end (Fig. 118–4). Immature glomeruli are often present. Existing glomeruli and tubules are enlarged. As the disease progresses, segmental sclerosis and hyalinosis of glomeruli are present. Tubular atrophy with interstitial fibrosis occurs. The renal artery is small.
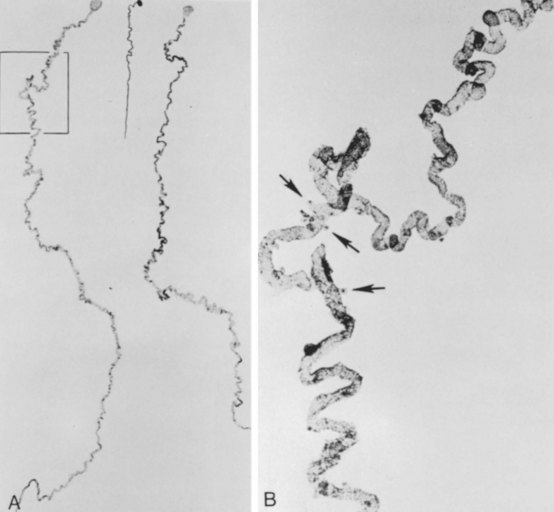
(From Fetterman GH, Habib R. Congenital bilateral oligonephronic renal hypoplasia with hypertrophy of nephrons [oligomeganephronia]. Am J Clin Pathol 1969;52:199–207.)
Treatment
Treatment is generally supportive and directed at maintaining normal biochemical balance, hemoglobin, and growth. High fluid intake and correction of salt loss and acidosis are the initial steps. Daily dietary protein should be limited to 1.5 g/kg during the stable phase (Royer et al, 1962). In addition, angiotensin-converting enzyme inhibitors may be of benefit in slowing progression of renal failure, even if the patient has a normal blood pressure. End-stage renal disease is managed by dialysis and transplantation. The allograft may come from a living related donor, because the disease is not familial.
Ask-Upmark Kidney (Segmental Hypoplasia)
Clinical Features
Most patients are 10 years of age or older at diagnosis. The disease is associated with severe hypertension, sometimes with headaches, either alone or together with hypertensive encephalopathy (Rosenfeld et al, 1973) and with retinopathy in half of the patients (Royer et al, 1971).
Proteinuria and some degree of renal insufficiency may be present if the disease is bilateral. Approximately half of the patients in the series described by Royer and colleagues (1971) had these signs at diagnosis. The disorder is usually unilateral, producing a small kidney with deep, narrow, segmental scars (slit-scar) found in the midzone. These slit scars are the classic radiographic finding. Although the scar can be detected with ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI), the presence of a deep scar is often overlooked.
Histopathology
The Ask-Upmark kidney is smaller than normal—12 to 35 g (Royer et al, 1971). Its distinctive feature is one or more deep grooves on the lateral convexity, underneath which the parenchyma consists of tubules resembling those in the thyroid gland. Usually, the hypoplastic segments are easily distinguished from adjacent areas. The medulla consists of a thin band, and remnants of the corticomedullary junction and arcuate arteries are seen. Arteriosclerosis is common, and juxtaglomerular hyperplasia may be seen (Bernstein, 1968; Meares and Gross, 1972; Kaufman and Fay, 1974; Arant et al, 1979) (Fig. 118–5).
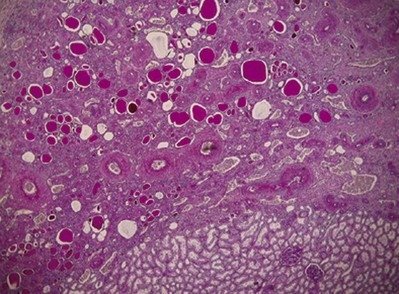
(From Babin J, Sackett M, Delage C, Lebel M. The Ask-Upmark kidney: a curable cause of hypertension in young patients. J Hum Hypertens 2005;19:315–6.)
Treatment
Abnormal renin secretion has been proposed as the cause of the hypertension; however, nephrectomy has been shown to normalize blood pressure regardless of plasma renin activities (Babin et al, 2005). In patients with unilateral disease, partial or total nephrectomy may control the hypertension (Royer et al, 1971; Meares and Gross, 1972). Failure of this measure suggests an unrecognized scar or generalized arteriosclerosis in the remaining kidney (Arant et al, 1979). Bilateral disease with renal insufficiency usually is managed medically, although dialysis and transplantation may be needed. Correction of reflux may prevent further renal damage but probably will have no effect on the hypertension.
Renal Hypodysplasia
According to Osathanondh and Potter (1964), posterior urethral valves may be associated with two types of renal hypodysplasia. In the less severe form, there are small, usually subcapsular cysts and nearly normal renal function. In the second form, the cysts are larger and more widely distributed, and numerous islands of cartilage are present. This form usually is associated with earlier onset and more severe obstruction and reflux (see Fig 118–2).
Cystic Diseases of the Kidney
The fundamental processes that are essential for the development and progressive enlargement of renal cysts include (1) proliferation of epithelial cells in segments of renal tubule, (2) accumulation of fluid within the expanding tubule segment, and (3) disturbed organization and metabolism of the extracellular matrix (Fig. 118–6).
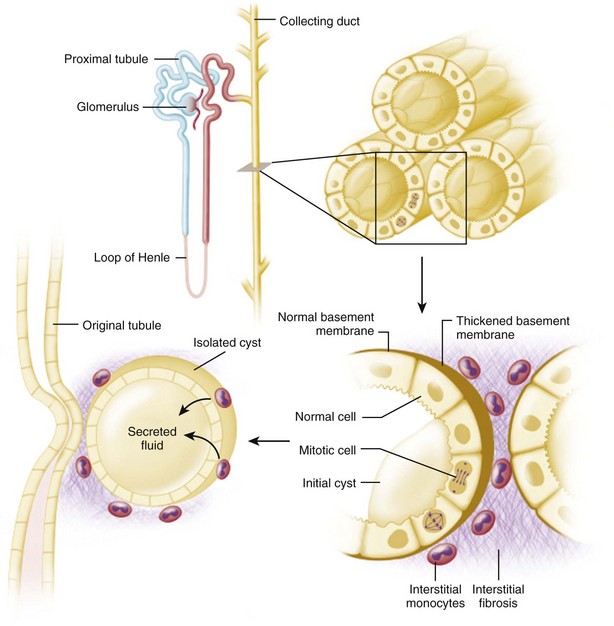
(From Brenner BM, editor. Brenner and Rector’s the kidney. 8th ed. Philadelphia: WB Saunders; 2007. p. 1428.)
An imbalance of the secretory and absorptive properties in proliferating epithelial cells leads to a net accumulation of fluid in otherwise normal renal tubules. Recent evidence indicates that, beyond the loop of Henle, tubule cells have the capacity to secrete solutes and fluid on stimulation with 3′,5′-cyclic adenosine monophosphate (cAMP) (Wallace et al, 2001). This secretory flux operates in competition with the more powerful mechanism by which sodium (Na+) is absorbed through apical epithelial Na+ channels (ENaC). Under conditions in which Na+ absorption is diminished, the net secretion of sodium chloride (NaCl) and fluid occurs.
Abnormalities of the extracellular matrix in and about renal cysts are seen in all cystic disorders. In the early stages of renal cyst development, changes in expression of collagen I and IV, metalloproteinase activators and inhibitors, integrins, and β-catenin may forecast a vital role for extracellular matrix remodeling in the pathogenesis of renal cysts. Recently, a hypomorphic mutation in the mouse laminin α5 gene was found to cause polycystic kidney disease (PKD) (Joly et al, 2006; Shannon et al, 2006).
Until recently, the mechanisms responsible for the abnormal differentiation and functional behavior of the epithelial cells that give rise to the cysts were largely unknown. Evidence now strongly suggests that a long-neglected structure, the primary cilium, is essential in maintaining epithelial cell differentiation. Structural and functional defects in the primary apical cilia of tubular epithelia may have a central role in determining cyst development and the abnormal differentiation and behavior of the cystic epithelium, and in various forms of human and rodent cystic diseases (Torres and Grantham, 2008).
Classification
In this chapter, the classification of renal cystic disease as outlined in 1987 by the Committee on Classification, Nomenclature, and Terminology of the AAP Section on Urology is used. The primary distinction is between genetic (“inheritable”) and nongenetic (“noninheritable”) disease. Table 118–1 gives an overview of characteristics associated with the various forms cystic disease.
Table 118–1 Cystic Diseases of the Kidney
| Inheritable |
| Noninheritable |
Inheritable Cystic Disease
Genetic (inheritable) cystic diseases can be classified based on their mode of transmission: autosomal dominant, autosomal recessive, X-linked, among others. Some of these disorders are caused by a single gene defect, some by an X-linked gene defect, and others by chromosomal defects. The modes of inheritance of these diseases and the specific gene or gene locus, where identified, are summarized in Table 118–2.

Autosomal Recessive (Infantile) Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease (ARPKD) is typified by relatively rapid, symmetrical, and bilateral enlargement of the kidneys in infants secondary to collecting duct cysts. It is invariably associated with some degree of congenital hepatic fibrosis (Zerres et al, 1988; Guay-Woodford and Desmond, 2003; MacRae Dell and Avner, 2003). ARPKD has been referred to as the “infantile” form in the past; however, the disease can present in adolescents and young adults, although significantly less frequently. ARPKD has a spectrum of severity, with the most severe forms appearing earliest in life. If it is not apparent at birth, the disease will become apparent later in childhood (up to age 13 years or, rarely, up to age 20 years).
The reported incidence of ARPKD varies from 1 in 10,000 to 50,000 live births (Zerres et al, 1988; Kaplan et al, 1989a). However, as many as 50% of affected newborns die in the first few days of life, making for a significantly lower incidence among children who live for at least 1 year. Of those infants who survive the neonatal period, approximately 50% are alive at 10 years of age (Kaplan et al, 1989a).
Genetics
Despite the clinical variability of ARPKD, it appears that only mutations of a single gene, named PKHD1 and located on chromosome 6 (6p12), are responsible for the disease. The gene produces a protein called fibrocystin (also known as polyductin) (Onuchic et al, 2002; Ward et al, 2002). This protein is a member of a larger family of proteins involved in regulation of cell proliferation and of cellular adhesion and repulsion. More specifically, dysfunction of this protein mediates cystogenesis through dysfunction of the primary cilia of renal epithelial cell. It is highly expressed in the kidney, with levels also present in the liver and pancreas. It is localized to the branching ureteric bud, collecting ducts of the kidney, and biliary ducts, consistent with the phenotype seen in ARPKD, and it is often absent in the patients with ARPKD (Wilson, 2004; Al-Bhalal and Akhtar, 2008).
Clinical Features
Affected children typically present in utero with enlarged, echogenic kidneys. Oligohydramnios is common because of the lack of normal urine production by the fetus. The infant often displays Potter facies and deformities of the limbs and may have respiratory distress as a consequence of pulmonary hypoplasia. The affected newborn usually has enormous, kidney-shaped, nonbosselated flank masses that are hard and do not transilluminate. In some cases, the kidneys are large enough to impede delivery. The infant’s serum creatinine and BUN concentrations are those of the mother’s at birth but soon begin to rise. Thirty to 50 percent of the affected individuals die shortly after birth as a result of uremia or respiratory failure (Kaplan et al, 1989c; Capisonda et al, 2003; Guay-Woodford, 2003). The earlier the age at which the disease is identified, the more severe the disease. No matter what the severity of the renal disease, all patients with ARPKD have liver involvement in the form of congenital hepatic fibrosis and vary in the degree of biliary ectasia and periportal fibrosis (Habib and Bois, 1973; Kissane and Smith, 1975).
Patients who survive the neonatal period have a milder form of the disease and are likely to survive into adulthood. Some diagnosed as neonates can live beyond age 3 or 4 years before going into renal failure (Cole et al, 1987; Avni et al, 2002). Hypertension and renal insufficiency are the major manifestations in surviving children, with liver disease becoming more prevalent in older patients. The exact cause of hypertension remains unclear. Consequences of chronic renal insufficiency (i.e., growth failure, anemia, osteodystrophy, etc.) become more apparent as the children age. Renal calcifications occur commonly in patients with ARPKD.
No association of ARPKD with renal neoplasms has been reported to date.
Histopathology
The kidneys are symmetrically enlarged (up to 20 times their normal size) and retain a reniform configuration. The parenchyma exhibits small subcapsular cysts, representing generalized fusiform dilations of the collecting tubules radiating from the medulla to the cortex with their long axis perpendicular to the renal surface (Bisceglia et al, 2006). Almost 100% of collecting ducts are affected in the most severe cases. The cortex is crowded with minute cysts (Fig. 118–7A). The renal pedicle is normal, as are the renal pelvis and ureter.

ARPKD affects both the kidneys and the liver in approximately inverse proportions (Torres and Grantham, 2008). The disease may be viewed as a spectrum ranging from severe renal disease and mild liver changes at one extreme to mild renal damage and severe liver disease at the other. The form with severe renal disease is the most common and the one typically seen at or near the time of birth. The form with less severe renal disease and more significant liver damage is present in older children. All children with ARPKD have lesions in the periportal areas of the liver (Habib and Bois, 1973; Kissane and Smith, 1975) (Fig. 118–7B). Congenital hepatic fibrosis is characterized by enlarged and fibrotic portal areas with apparent proliferation of bile ducts, absence of central bile ducts, hypoplasia of the portal vein branches, and sometimes prominent fibrosis around the central veins. Bulbar protrusions from the walls of dilated ducts also occur, and bridges sometimes form. This malformation has been found to occur occasionally as an isolated event (Caroli disease), but most often it is associated with ARPKD (Torres and Grantham, 2008).
Evaluation
The diagnosis may be suspected from in utero ultrasound examination and may be associated with oligohydramnios, a finding secondary to low urinary output. In both fetus and newborn, ultrasonography identifies bilateral, very enlarged, diffusely echogenic kidneys, especially when compared with the echogenicity of the liver (Fig. 118–8A). The increased echogenicity is due to the presence of numerous microcysts (created by tightly compacted, dilated collecting ducts) that result in innumerable interfaces. Compared with normal newborn kidneys, in ARPKD, the pyramids are hyperechogenic because they blend in with the rest of the kidney, and the kidneys typically have a homogeneous appearance (see Fig. 118–8A). In ADPKD, the cysts, if apparent in newborns, are usually diffuse and large (see Fig. 118–8B). Also included in the differential diagnosis is severe bilateral hydronephrosis (the kidneys are enlarged with hypoechogenic calyces), multicystic kidney (hypoechogenic cysts lie within a nonreniform mass that has very little parenchyma), sporadic glomerulocystic kidney disease, bilateral mesoblastic nephroma, Wilms tumor, and bilateral renal vein thrombosis. If the diagnosis is in doubt, computed tomography is valuable because it is more sensitive to inhomogeneity (and therefore to tumor) within abdominal masses.

Considerable overlap in clinical presentation and imaging findings can occur between ADPKD and ARPKD. Occasionally, a newborn with severe ADPKD can also have enlarged, homogeneously hyperechogenic kidneys. Age at onset can overlap, and typically, when ADPKD manifests at birth, cysts are apparent on the sonographic image. Macrocysts are rare in newborns with ARPKD but do increase in frequency as the child gets older, sometimes producing an appearance similar to that of the dominant disease. Cysts less than 1 cm appear more often than large cysts (Avni et al, 2002) (Fig. 118–9). Hepatic fibrosis can rarely occur in patients with ADPKD, as can brain aneurysms with ARPKD (Cobben et al, 1990; Neumann et al, 1999). The correct diagnosis of ARPKD depends on the overall clinical data: positive family history with a recessive mode of inheritance, a positive liver biopsy, and the lack of the extrarenal malformations often associated with other renal cystic diseases.
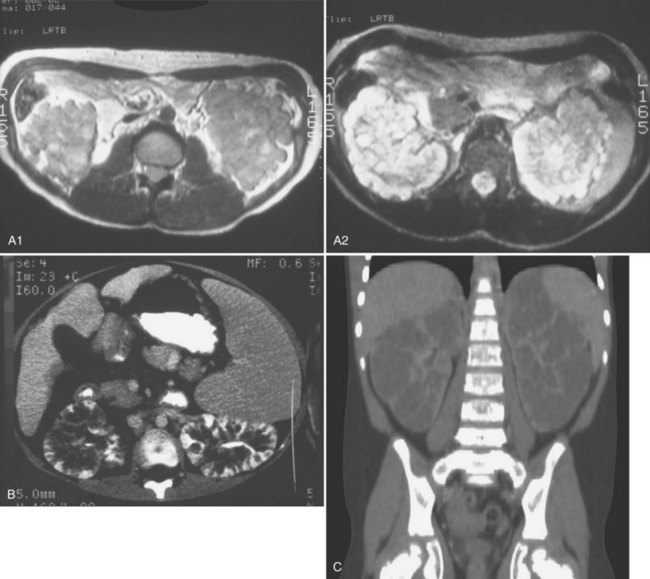
(A, Courtesy of Walter Berdon, MD. B, Courtesy of Marta Hernanz-Schulman, MD.)
Autosomal Dominant (Adult) Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease is by far the most common inheritable form of renal cystic disease, with an incidence of approximately 1 in 400 to 1000 live births (Iglesias et al, 1983; Grantham, 1996). It is an important cause of renal failure, accounting for 7% to 15% of patients who receive hemodialysis (Hildebrandt, 1995; Grantham, 1996; Wilson, 2004). The trait theoretically has a 100% penetrance, and, on average, because it is transmitted in an autosomal dominant fashion, 50% of an affected individual’s offspring will likewise be affected. Although a positive family history is one of the major criteria for the diagnosis of ADPKD, 10% of cases occur sporadically. Ninety-six percent of affected persons will manifest the disease clinically by age 90 years (Gabow, 1991).
Although most cases are identified between the fourth and fifth decades of life, the condition has been reported in newborns and infants (Proesmans et al, 1982). All affected individuals manifest the disease (although not necessarily symptomatically) if they live long enough, but renal failure is seldom seen before the age of 40 years, unless the disease manifests during infancy, in which case it is much more aggressive.
A number of associated anomalies are common, including cysts of the liver, pancreas, spleen, and lungs; aneurysms of the circle of Willis (berry aneurysms); colonic diverticula; aortic aneurysms; and mitral valve prolapse (Table 118–3).
Table 118–3 Comparison of Autosomal Recessive and Autosomal Dominant Polycystic Kidney Disease
| AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE | AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE | |
|---|---|---|
| Gene defect | Chromosome 6 | Chromosome 4 and 16 |
| Incidence | 1: 5000-40,000 | 1: 500-1100 |
| Usual age at clinical presentation | Perinatal | Third to fifth decades |
| Typical ultrasonographic appearance of kidneys | Symmetrically enlarged, homogeneous, hyperechogenic | Large, cystic kidneys, sometimes asymmetrical |
| Histology | Collecting duct ectasia; cysts derived principally from collecting duct | Microcysts and macrocysts derived from the entire nephron |
| Liver | Always with congenital hepatic fibrosis but of varying severity | Cysts, mostly in adults |
| Other organ system(s) involvement | None | Intracranial (berry) aneurysms, colonic diverticuli; mitral valve prolapse; cysts of other organs (seminal vesicle, arachnoid membrane, pancreas) |
Genetics
There are two major genetic forms of ADPKD caused by mutation in the genes PDK1 and PDK2 (Reeders et al, 1985; Breuning et al, 1987; Ryynanen et al, 1987; Pieke et al, 1989). PDK1 has been localized to the short arm of chromosome 16, with its gene product being polycystin-1. Mutations in the PDK1 gene account for 85% of ADPKD cases. These patients typically have a more rapidly progressive form of the disease, with cysts usually developing by the age of 20 years (64% of children have cysts by the age of 10 years and 90% by the age of 20 years) and ESRD occurring in their 50s (Chakraborty and McHugh, 2005). The PKD2 gene has been mapped to the long arm of chromosome 4 (Peters et al, 1992), and it encodes the protein polycystin 2. PDK2 mutations account for about 15% of ADPKD cases, and in these patients, the disease progresses more slowly. ESRD usually does not occur until these patients are in their 70s (Hateboer et al, 1999). The presence of a third locus (PKD3) is now accepted as the cause of disease in a very small percentage of patients who have been found to have neither a PKD1 nor a PKD2 gene defect (Dauost et al, 1993). When either the PKD1 or the PKD2 gene is abnormal, cystic kidneys can develop. Because defects of PKD1 and PKD2 manifest similarly, it has been suggested that the gene product of each is involved in the same pathway and that an abnormality of either product results in similar manifestations of the disease (Qian et al, 1996).
Every cell of the nephron and collecting duct has the PDK1 or PDK2 mutation; however, only 1% to 2% of these glomerular units are affected by cyst formation. Only those nephrons that undergo a disruption of a second allele undergo cystic enlargement. This is the “second hit” of the Knudson theory, which has been proposed to explain the focal nature of the cysts (Knudson, 1991). In this model, a mutated PKD1 (or PKD2) gene is inherited from one parent, and a wild-type gene is inherited from the unaffected parent. During the lifetime of the individual, the wild-type gene undergoes a somatic mutation and becomes inactivated. Loss of heterozygosity owing to somatic mutations of the PKD1 and PKD2 genes has been identified in the cells lining the cysts in both the kidney and liver (Qian et al, 1996; Watnick et al, 1998). The phenomenon of genetic anticipation is seen as well; it is manifested by progressively earlier presentation and increased severity in subsequent generations of patients with ADPKD (Fick et al, 1994; Zerres et al, 1994). Genetic testing for PDK1 and PDK2 are commercially available through polymerase chain reaction amplification and direct DNA sequencing.
Pathogenesis
The PDK1 and PDK2 proteins, polycystin 1 (PC1) and polycystin 2 (PC2), are transmembrane proteins that in all likelihood form a functional complex. When working properly, PC1 and PC2 inhibit cell proliferation by several pathways. Polycystin 1 functions as a mechanoreceptor located on the primary cilium of renal tubular cells. These cilia project from the surface of the cell into the lumen of the ducts and tubules. Mutations that block the assembly of these cilia are known to cause many cystic diseases of the kidney. The products of the ADPKD, ARPKD, and nephronophthisis genes are at least partially localized to these primary cilia. Polycystin 1 is linked to polycystin 2, which contains a calcium (Ca++) channel. When the mechanoreceptor of polycystin 1 is stimulated by calcium-containing urine flowing through the tubule, the calcium channel of polycystin 2 opens and calcium enters the cell (Nauli et al, 2003; Braun, 2009). This process regulates the proliferative state of the renal tubular cells through a variety of signaling pathways (cAMP, extracellularly regulated kinase [ERK], mammalian target of rapamycin [mTOR], etc.). The cilia likely serve as organization centers for this signal transduction (Pazour, 2004).
Increased levels of cAMP have been noted in multiple animal models of ADPKD, and biochemical processes that alter cAMP appear to affect the presence and size of renal cysts. For example, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products, such as theophylline, interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD, and increase cyst formation in canine kidney cell cultures (Mangoo-Karim et al, 1989; Belibi et al, 2002). The abnormal response to cAMP is directly linked to alterations in Ca++ channel activity and results in the abnormal response of cyst-derived cells. This has also been shown to be regulated by cAMP stimulation of the mitogen-activated protein kinase/extracellularly regulated kinase (MAPK/ERK) signaling pathways, which leads to abnormal cellular proliferation of the PKD renal epithelial cells.
Upregulation of the vasopressin V2 receptor has also been shown to increase cAMP levels. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function (Gattone et al, 2003; Wang et al, 2005). Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats (Nagao et al, 2006). Polycystic kidney (PCK) animals with no vasopressin activity had virtually no cAMP or renal cysts, whereas animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease (Wang et al, 2008; Braun, 2009). Finally, the absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts (Shillingford et al, 2006). The mTOR system can be blocked by rapamycin, which has been shown to slow PKD progression in rats (Wahl et al, 2006).
Clinical Features
Phenotypes associated with ADPKD are highly variable in penetrance. This variability related to the severity of renal disease can range from fetal demise and neonatal death to adequate renal function into old age. Characteristics of the associated liver disease and extrarenal manifestations can also be quite variable. PDK1 and PDK2 gene mutations are obviously critical, but intrafamilial variability also indicate that genetic background and environmental factors can also affect ultimate clinical expression of the disease (Rossetti and Harris, 2007). Now that the families of ADPKD patients are being screened by ultrasonography, large numbers of asymptomatic children with renal cysts are being identified before full-blown disease develops. Bilateral renal involvement is the usual presentation, but 17% of cases are asynchronous/asymmetrical, especially in children. These extreme forms of asymmetry are referred to as “unilateral ADPKD” with the involvement in the contralateral kidney becoming apparent at a much older age (Strand et al, 1989; Bisceglia et al, 2006).
Typically, signs or symptoms first occur between the ages of 30 and 50 years (Glassberg et al, 1981). These include microscopic and gross hematuria, flank pain, gastrointestinal symptoms (perhaps secondary to renomegaly or associated colonic diverticula), renal colic (secondary either to clots or stones), and hypertension. Microscopic or gross hematuria is seen in 50% of patients, and, in 19% to 35%, it is the presenting symptom (Milutinovic et al, 1984; Delaney et al, 1985; Zeier et al, 1988; Gabow et al, 1992). Because patients with ADPKD have increased renal mass, erythropoietin levels are increased, making anemia unusual even when ESRD is present (Gabow, 1993).
Pain (flank and/or abdominal) is the most common presenting symptom in adults. This results from a number of possible factors: mass effect (cysts impinging on abdominal wall or neighboring organs), bleeding into the cysts, urinary tract infection (including infected cysts), and nephrolithiasis. Twenty to 30 percent of patients with ADPKD develop stones (Fick et al, 1994), and these are treated by conservative means if possible (i.e., urine alkalinization, spontaneous stone passage, extracorporeal shock wave lithotripsy). Uric acid stones and calcium oxalate stones are equally prevalent. The finding of hydronephrosis, which helps make the diagnosis of stones, may not be as useful in ADPKD patients because of the number of cysts camouflaging the findings (Choyke et al, 1995). Urinary tract infections are more frequent in female patients. Cyst aspiration for both diagnostic and therapeutic reasons should be considered in the setting of suspected cyst infection.
As blood pressure screening has become more widespread, hypertension has become a very common form of presentation (Zeier et al, 1988). Hypertension is present in roughly 50% of patients 20 to 35 years old having ADPKD with normal renal function. Virtually 100% of patients with ESRD will have hypertension (Kelleher et al, 2004
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
Minimally Invasive and Endoscopic Management of Benign Prostatic Hyperplasia
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


