Fig. 2.1
Autosomal dominant polycystic kidney disease . The kidney is massively enlarged with innumerable cysts
Histologically, the cysts are lined by a single layer of cuboidal or flattened epithelial cells (Fig. 2.2). Focal cellular proliferations and small papillary formations may develop along the cysts lining (Fig. 2.3). The intervening renal parenchyma in the early stages of cyst formations can be normal. Both tubular and glomerular cysts can be present [23]. In the advanced stage, the parenchyma is attenuated in between cysts and exhibits interstitial fibrosis, tubular atrophy, glomerular sclerosis, calcifications, and lymphocytic infiltrates. About two-thirds of the rare renal tumors arising in ADPKD exhibit a papillary phenotype (Fig. 2.4).
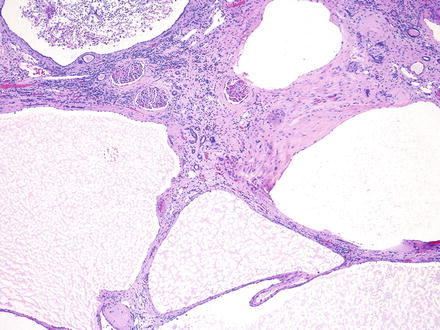
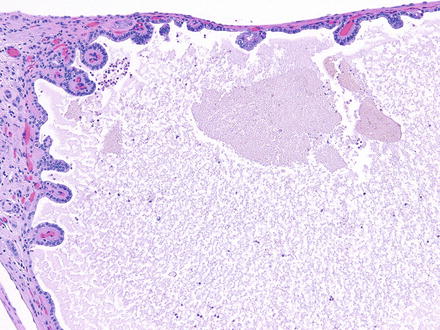
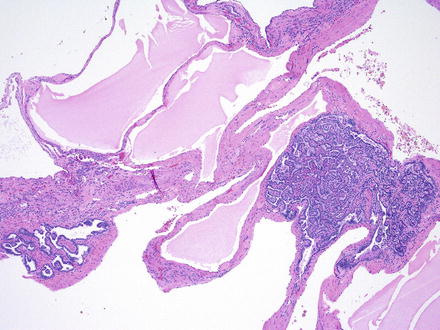
Fig. 2.2
The cysts in ADPKD are unilocular with simple flat to cuboidal epithelial lining . The renal parenchyma exhibits interstitial fibrosis and chronic inflammation
Fig. 2.3
Simple papillary formations in the cyst lining of ADPKD
Fig. 2.4
Papillary adenomas arising in ADPKD
Clinical Presentation
The phenotype of ADPKD can be highly variable despite the known genetic inheritance patterns caused by mutations in PDK1/ PDK2 . Aside from the “second-hit” theory of cyst formation, the genetic, hormonal, and environmental backgrounds of patients with ADPKD appear to affect disease manifestation. Characterization of these external factors is the subject of ongoing research [24].
Disease signs and symptoms usually begin between the 3rd and 5th decades of life for those with the PDK1 gene mutation. The main presenting symptoms include flank pain, microscopic and/or gross hematuria, gastrointestinal discomfort, renal colic, and hypertension [25–28]. Up to 43 % of patients experience at least one episode of gross hematuria and 25 % report more than six episodes. Recurrent episodes of gross hematuria have been associated with an accelerated decline in renal function [29].
Pain is the most common presenting symptom, and its etiology is multifactorial. The size of the cysts and the location of the cysts with relation to other organs, the abdominal wall, and the renal collecting system can affect the type of pain a patient experiences. In full-blown ADPKD, each kidney can weigh several kilograms and occupy a large volume within the abdomen, displacing other organs. Furthermore, complications such as hemorrhage into cysts, infected cysts, urinary tract infections, and nephrolithiasis can make appropriate diagnosis and treatment challenging. Up to 30 % of patients with ADPKD develop nephrolithiasis, though identifying obstruction and hydronephrosis can be difficult when multiple large cysts are present [30–32].
Hypertension is almost universally present in patients with ADPKD [28]. About half of all patients in their 2nd or 3rd decade will have hypertension with normal renal function, with an increasing incidence to 100 % by the time they acquire ESRD [33]. Several mechanisms have been proposed to explain the high prevalence of hypertension in this population, most of which involve the renin-angiotensin-mediated pathway activated by ischemic glomerular units. It is postulated that cyst growth causes stretching of intrarenal arteries and subsequent ischemia of the zones dependent on this vasculature [26]. As further ischemia occurs, renal dysfunction ensues and leads to worsening hypertension, which in turn leads to vascular remodeling and further decline in renal function.
Diagnosing ADPKD just from declining renal function is rare, as most renal function is preserved until the 4th–6th decade of life. However, the presence of enlarging cysts and/or enlarging kidneys is proportionately related to the decline in renal function. As such, monitoring kidney size may help alert clinicians of impending functional decline [34].
Extrarenal Manifestations
Hepatic cysts are the most common extrarenal manifestation of ADPKD, and virtually all patients will exhibit some hepatic cyst formation by their 5th decade [35]. The hormonal influence of estrogen has been found to increase cyst formation and growth; the incidence of hepatic cysts is therefore higher in females than males [31, 36]. Although usually asymptomatic, hepatic cysts can rarely be responsible for mass effects, cyst hemorrhage, and portal hypertension from hepatic fibrosis, though portal hypertension is almost uniquely a finding of ARPKD [37].
Intracranial aneurysms (ICAs) , or berry aneurysms , can be an immediately life-threatening extrarenal manifestation of ADPKD. 10–30 % of ADPKD patients exhibit ICAs, and the risk of aneurysm rupture increases significantly with poorly controlled hypertension [38, 39]. There is an increasing incidence with age; hence, it is proposed (though not universally practiced) that all patients older than 45 years of age get regular screening for ICAs [40]. ICAs form relatively quickly and are caused by a defect in the elastic lamina of vessel walls, allowing outward bulging that either ruptures or stabilizes due to collagenous remodeling. It is thought that ICAs <7 mm are less likely to grow or rupture, but since ADPKD results in wall protein deficiency (polycystins 1 and 2 are expressed in vascular smooth-muscle cells), the regular rules for ICAs may not apply [41]. ICA formation is a hypertension-independent event; thus, proper control of blood pressure is critical in preventing rupture [42]. The role of MRI screening for berry aneurysms is somewhat controversial, but evidence does seem to suggest that patients with a positive family history of ruptured ICAs and those with a PDK1 genotype are at highest risk of mortality and should therefore be considered for earlier screening. Coronary and abdominal aortic aneurysms have also been associated with ADPKD; screening should be considered, especially for those on hemodialysis [43].
Diagnosis/Evaluation
Most patients will present after their 2nd or 3rd decade, but as mentioned previously, pediatric cases do arise. In these cases, radiographic characteristics may mimic ARPKD, and it may take time for macrocysts to appreciably develop. In these patients, an evaluation of family history is critical to making the correct diagnosis. Genetic testing should be discussed with members of the patient’s family who are at risk for carrying the disease. All the usual implications of genetic counseling apply in ADPKD. The benefits of allowing for family planning and early intervention should be carefully weighed against the harms of psychological and psychosocial stress induced by knowing the diagnosis . Of particular interest in family genetic counseling is identifying related individuals who do not have the disease and can act as living-related organ donors. Pregnancy in patients with ADPKD can also potentially lead to more rapid progression toward ESRD and enlargement of hepatic cysts from estrogen effects. However, fertility itself is preserved, especially if renal function is normal [45].
In the index patient, one can use simple radiographic findings to guide the diagnosis. Renal ultrasound can be used with high sensitivity to guide diagnosis using the following criteria: the presence of at least two renal cysts (unilateral or bilateral) in individuals at risk and younger than 30, the presence of at least two cysts in each kidney for patients aged 30–59, and the presence of at least four cysts in each kidney in those above the age of 60 [46]. This results in a sensitivity of 100 % for those older than 30 years of age. However, caution should be exercised in patients younger than 30 with suspected PDK2 mutations because ultrasound is only about 67 % sensitive in these cases; hence, further DNA linkage analysis would be prudent [47]. Occasionally, patients will present with predominantly extrarenal manifestations or with incidentally discovered cysts without a family history of disease. The diagnosis of ADPKD can be established with the presence of two or more renal cysts along with a polycystic liver, an ICA, or pancreatic cysts [45].
The use of axial imaging is not strictly necessary to diagnose ADPKD. However, CT and MRI can be useful adjuncts in the monitoring of disease progression or for assisting in the diagnosis of complicated presentations (Fig. 2.5). Contrasted CT imaging is useful in assessing hemorrhage within a cyst by demonstrating a predictable rise in contrast density [30]. Newer advances in MRI computing technology have the ability to segment individual renal cysts in ADPKD kidneys and provide a quantitative indicator of severity in early and moderate stages of disease [48]. MRI can also be an important tool to evaluate cystic lesions or hemorrhage in patients with advanced renal dysfunction who cannot receive the contrast load required for appropriate CT imaging.
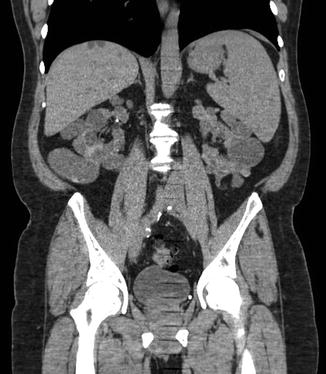
Fig. 2.5
Coronal CT scan view of a patient with ADPKD. Note the derangement of normal renal architecture by innumerable large renal cysts. Some small liver cysts are also noted on this view
Nocturia can sometimes be an early sign of decreased renal concentrating ability, and non-nephrotic proteinuria is present in ~33 % of patients with ADPKD. Recent advances in urinary proteomics have identified patterns that can aid in diagnosis and risk stratification with a sensitivity of 85 % and specificity of 94 %, with some biomarkers able to predict height-adjusted total kidney volume [49]. Assessing total kidney volume (TKV) is an important step in evaluating and monitoring these patients, as TKV has been confirmed as a clinically meaningful surrogate marker in ADPKD [50]. This metric was established by the Consortium for Radiological Imaging in Studies of Polycystic Kidney Disease (CRISP Study) , which helped identify markers of disease progression. It was found that patients with higher rates of kidney enlargement and overall larger TKV have a more rapid decline in kidney function and should be monitored more closely [51].
In patients with a family history of ruptured ICAs, cranial MRI is appropriate to identify asymptomatic patients with ICAs . Some authors recommend screening all ADPKD patients above the age of 45 due to a prevalence of ICAs of up to 22 % in this age group. Older patients generally have an increased risk of rupture, especially in the setting of poorly controlled hypertension [40]. Although ICA rupture is considerably rarer in younger patients, identification and early management of an ICA in a younger patient would lead to the greatest benefit in life expectancy. However, it remains unclear if the natural history of aneurysm formation, growth, and rupture-risk parallels that of the general population; and thus more data are still required before definitive radiographic guidelines can be established to screen for ICAs [41, 52].
In some patients with recurrent upper tract or cyst infections, prompt treatment is imperative but can be hindered by difficulty in evaluation. Definitive diagnosis requires evaluation of cyst fluid, but a probable diagnosis can be made if all four of the following criteria are met: temperature >38C for >3 days, loin or liver tenderness, C-reactive protein plasma level of >5 mg/dL, and no evidence for intracystic bleeding on CT. Positron-emission tomography (PET) after intravenous injection of 18-fluorodeoxyglucose (PET/CT) has also been studied as a more reliable imaging modality when evaluating for the presence of pyocysts [53].
Sequelae
Cyst infections, UTIs, nephrolithiasis, chronic pain, hematuria, progression to ESRD, and a possible increased risk of renal malignancy are all sequelae for which these patients should be monitored and counseled.
The progression to end-stage renal disease (ESRD) is an important clinical outcome that must be evaluated in all patients once the diagnosis of ADPKD is established. eGFR declines rapidly in the decade preceding ESRD, at a rate of 4.4–5.9 ml/min/year; and a 30-year retrospective analysis from the United Kingdom demonstrated that a decline in eGFR can be seen as early as 20 years prior to ESRD, affording an earlier ability to stratify for patients that will have a more aggressive decline in renal function [54]. A combination of serum chemistries and radiographic follow-up is the ideal method to monitor this decline, and management by Nephrology is mandatory to preserve renal function for as long as possible. About half of patients with ADPKD will eventually progress to ESRD.
There is a small risk of hepatic dysfunction in these patients, though the mechanism is likely not directly related to ADPKD . Hepatic cysts are usually asymptomatic; but in rare cases, such as women in whom cyst growth can be greater, direct compression of the portal vein can result in portal hypertension [37]. Portal hypertension can also be a symptom of primary hepatic fibrosis . Unlike in the autosomal recessive disease, ADPKD is not thought to be directly associated with primary hepatic fibrosis. Nonetheless, there can be a concomitant diagnosis of hepatic fibrosis, especially with a perinatal diagnosis of ADPKD.
The incidence of renal cell carcinoma (RCC) in patients with ADPKD has been a matter of debate in the literature. The incidence of renal adenomas is 1 in 4–5 patients, similar to the incidence seen in patients with cystic disease related to dialysis [9]. The combination of ADPKD and dialysis is thought to raise the incidence of RCC up to 2–3 times the frequency seen in ESRD patients without ADPKD [22, 55]. However, ADPKD patients without ESRD appear to have a similar RCC incidence as the general population. In a retrospective analysis of 240 ADPKD patients who underwent renal surgeries, 12 (5 %) patients presented with malignant lesions. Two-thirds of these patients had undergone dialysis prior to surgery. Of the malignant tumors identified, 63 % were papillary RCC, 31 % were clear cell RCC, and 6 % had papillary noninvasive urothelial carcinoma [22]. Other characteristics associated with ADPKD patients who develop RCC include diagnosis at a younger age, bilateral tumors (12 % vs. 1–5 % in the general population), multicentric tumors (28 % vs. 6 % in the general population), and sarcomatoid histologic features (33 % vs. 1–5 % in the general population) [56].
Importantly, retrospective series studying the incidence of RCC in ADPKD patients are limited by the small numbers of patients who actually develop malignant lesions. However, it seems apparent that the combination of ADPKD and ESRD with at least 1 year of dialysis does confer a greater risk of developing malignant lesions [55]. Unfortunately , a variety of clinical features of the disease may mask or prevent diagnosis of malignant lesions, including cystic distortion impairing imaging quality, cystic hemorrhage, and proteinaceous debris, all of which are common findings in regular ADPKD kidneys.
Treatments
The common theme for all treatments is to prevent secondary or downstream effects of the disease, such as hypertension, pain, infection, and ESRD. Appropriately controlling hypertension is likely the most important factor in preventing future morbidity. As discussed, uncontrolled hypertension is a harbinger for rapidly declining renal function, unstable berry aneurysms, and significant cardiovascular disease. The use of antihypertensives, specifically ACE inhibitors , appears to be the most effective way to manage blood pressure, especially when considering that hypertension from ADPKD is most likely a renin-based phenomenon. The recently published HALT-PKD study evaluated the combined use of renin-angiotensin agents and blood pressure targets on total kidney volume growth, estimated GFR, and urinary albumin excretion [19, 57]. Findings suggest that the combination of an ACE inhibitor (lisinopril) + angiotensin receptor blocker (telmisartan) had no significant benefit versus standard blood pressure management, which normally includes just an ACE inhibitor. Interestingly, the lower blood pressure group (95/60 to 110/75 mmHg), as compared to the standard blood pressure group (120/70 to 130/80 mmHg) had a slower increase in total kidney volume and greater reduction in urinary albumin secretion, but did not affect estimated GFR [58]. These findings seem to suggest that tight blood pressure regulation in ADPKD patients may slow the progression of disease.
Many patients will inevitably require dialysis for ESRD . Obviously, patients who require unilateral or bilateral nephrectomy , such as for mass effect or tumors, are at highest risk for needing renal replacement. However, a gradual progression to ESRD can be predicted for many symptomatic patients, and early planning and discussion with patients can help when decisions are required regarding renal replacement. Guidelines for renal transplant vary by geographic location, but should be considered when appropriate.
Clinicians should be aggressive in treating stones and infections. Patients with ADPKD almost universally have chronic pain, which can complicate the diagnosis and clinical presentation of treatable pathologies. Although these patients almost universally have chronic pain, there should be a high risk of suspicion for obstruction (from stones or cyst compression) when patients present with acute pain. Prompt treatment of the offending pathology can help save renal function. Pain should be managed carefully in this population, since opioid dependence is common and even surgical intervention may not completely alleviate symptoms. Nephrotoxins such as nonsteroidal anti-inflammatories (NSAIDs) should be avoided as much as possible.
The prevalence of kidney stones in this patient population appears to be greater than in the general population. ADPKD patients more prone to stone formation are those with lower eGFR and more/larger renal cysts. Metabolic evaluation in ADPKD stone formers tends to demonstrate lower urinary volumes and lower levels of urinary phosphate, magnesium, potassium, and citrate. Prevention of stone formation in these patients requires attention to metabolic factors (increasing stone inhibitors such as water, magnesium, and citrate) and structural factors (preventing large cyst formation) [32].
Some patients, especially women, are at high risk of developing upper tract infections and should be treated aggressively with IV antibiotics for pyelonephritis . Occasionally, infections can be harbored in noncommunicating cysts and will need to be surgically drained or percutaneously aspirated.
Very large cysts causing clinical symptoms can be managed in a variety of ways. They occasionally require surgical unroofing (usually done laparoscopically) or percutaneous aspiration with chemical ablation [59]. Nephrectomy may be an appropriate step in patients with severe pain who have already developed ESRD [60]. Open surgical excision via a lumbodorsal or abdominal midline incision has been described as an effective way to remove very large kidneys. However, improvements in laparoscopic techniques have allowed laparoscopic surgeons to effectively and safely perform a nephrectomy on many of these patients [61].
There are a variety of medical therapies being tested to control the primary disease process. Many of these treatments are aimed at decreasing intracellular cAMP, which is produced as a result of overproduction of vasopressin in ADPKD patients [62–65]. cAMP contributes to cyst and kidney enlargement and subsequent renal dysfunction. Tolvaptan, a V2 receptor antagonist that downregulates cAMP production, was shown to slow the increase in TKV, but unfortunately did not improve eGFR [66].
Somatostatin analogs , such as octreotide, have been found to inhibit cAMP pathways that lead to fluid secretion into cysts. A randomized clinical trial studying the effectiveness of somatostatins showed that it slowed kidney and liver growth, improved patient health perception, and was well tolerated [67].
The mammalian target of rapamycin (mTOR) inhibitors sirolimus and everolimus have each been studied in RCTs [68, 69]. Both had a relatively short follow-up period (~2 years), making it difficult to determine the long-term effect of mTOR inhibition. Nonetheless, everolimus did appear to decrease total kidney growth compared to placebo, though the effect on eGFR decline was unclear [68, 70]. Several antineoplastic drugs have been tested, including lonidamine and curcumin, with the idea that abnormal proliferative pathways in ADPKD kidneys can be generically targeted. Though these drugs have met with limited experimental success, it awaits to be seen if they play a role in the medical management of ADPKD in the future. Other possible therapies have focused on the cystoprotein complexes involved in aberrant signaling cascades, such as the Src pathway . Metformin , a commonly used oral diabetic agent, may activate AMP-activated protein kinase, thereby inhibiting CFTR and mTOR pathways that are responsible for downstream cyst formation [19]. Inhibition of HSP90 has been shown to induce degradation of many ADPKD-relevant genes in mouse models. This was associated with a decrease in cyst formation and improved renal function, which is a promising lead for future investigation [7].
Unfortunately, no current medical therapies have shown a significant effect on the overall progression of disease. For this reason, it is essential that patients be monitored for secondary effects of the disease, such as hypertension and other above-mentioned pathologies that can decrease the severity of consequent morbidity.
Prognosis
Though the disease has a very heterogeneous phenotypic presentation, patients who are managed appropriately can have excellent long-term health. In a 30-year natural-history retrospective analysis of patients with ADPKD, investigators were able to demonstrate that the rate of eGFR change can predict progression to ESRD as much as 10 years before onset. It is therefore critical to continually assess renal function once the diagnosis of ADPKD is established. The analysis also showed that the rate of eGFR change can be predicted by mean kidney length and patient’s age at the time of diagnosis [54]. Patients diagnosed at a later age have a slower rate of renal function decline than do patients who are diagnosed young, which supports the idea that early manifestation of the disease may indicate more aggressive clinical progression.
Monitoring for hypertension, infections, and, in select patients, intracranial vascular aneurysms is key to long-term health. A multidisciplinary approach should be used when managing these patients, including a primary care physician, Nephrologist, and Urologist . Patient education about the disease process and prognosis can greatly aid in early identification of treatable pathologies.
Autosomal Recessive Polycystic Kidney Disease
Background
Autosomal recessive polycystic kidney disease (ARPKD) is a rare but severe congenital hepatorenal disease that affects an estimated 1 in 20,000 live births [71]. It is a neonatal disease characterized by symmetric, massively enlarged kidneys and congenital hepatic fibrosis that presents with varying clinical severity. Typical disease presentation occurs in neonates and includes fetal oligohydramnios, massively enlarged kidneys, and “Potter” sequence abnormalities, such as pulmonary hypoplasia and severe respiratory insufficiency [72]. Perinatal death occurs in 30–50 % of affected newborns [73, 74]. For those who survive the perinatal period, long-term survival depends on the degree of renal and hepatic dysfunction. One-year survival has been reported as high as 92–95 % for those who survive the first month of life [72]. Systemic hypertension, pulmonary hypertension, impaired nutrition from GI mass effect, pulmonary insufficiency, and neurocognitive dysfunction are all sequelae that can greatly affect the morbidity associated with the disease in survivors [75].
ARPKD is thought to be in a similar class as a host of other hepatorenal fibrocystic diseases , such as ADPKD, Joubert syndrome, Bardet-Biedl syndrome, glomerulocystic disease, renal-hepatic-pancreatic dysplasia, and Zellweger syndrome, just to name a few [75]. The common thread is that they all have defects localized to primary cilia/basal body structures, which in turn appears to result in similar hepatorenal dysfunction.
The implicated gene, PKHD1 , is now required for the diagnosis of ARPKD, especially given its overlap with other syndromes. The recent discovery of this gene has allowed clinicians to better understand the heterogeneity of disease presentation, and is helping elucidate better molecular targets for future treatments [76, 77]. The improved genetic understanding of the disease has also changed the psychosocial impact the diagnosis has on families. Since families can now be tested for the implicated gene mutations, they can face difficult decisions with regard to future reproduction. As such, a multidisciplinary approach to disease management is critical when a patient is diagnosed with ARPKD.
Genetics/Pathogenesis
ARPKD is transmitted in the usual autosomal recessive pattern. Although newer genetic testing can identify the majority of mutant alleles for diagnostic purposes, appropriate pedigrees should be constructed for at least three generations to identify the carriers of the diseased alleles. ARPKD is caused by mutations to PKHD1, a large gene with a very complex splicing pattern that is located on chromosome 6p21.1-p12 [76, 77]. The gene product is fibrocystin/polyductin (FPC) , a transmembrane protein that is expressed predominantly in renal collecting ducts, the thick ascending loops of Henle, pancreatic tissues, and hepatic bile duct epithelia [4, 76, 78].
Although the exact function of FPC is unclear, it is known to localize to ciliary/basal body structures and apical membranes, which associates it with other diseases caused by ciliary defects that manifest in hepatorenal fibrocystic disease . The exact molecular mechanism of pathogenesis is unclear, though the complex does interact with polycystin-2, one of the ADPKD genes.
PKHD1 is a long gene that is susceptible to a large variety of mutations along its entire length, the majority of which are missense mutations or truncations. Over 300 mutations are currently known, though there are likely more that spontaneously form [75]. Given the heterogeneity of mutations, most patients with ARPKD are actually compound heterozygotes and carry two different mutant alleles, contributing to the large variation in disease phenotypes. Many patients are found to have novel mutations when genetic analysis is completed, making interpretation and prognostication of disease severity difficult.
Pathology
Both kidneys in lethal neonatal form are massively enlarged although their reniform configuration is generally maintained [79, 80]. Numerous cysts derived from the collecting ducts are present in the medulla and inner cortex that impart a spongy appearance. These cysts in neonates are elongated (in contrast to the rounded cysts in ADPKD) and are characteristically oriented perpendicular to the renal cortex (“radial rays”). The cysts are lined by small cuboidal epithelial cells. Most cysts represent ectatic collecting ducts, although glomerular cysts may also be present [23]. In infantile and childhood cases, the involved kidneys can be smaller with variable round to elongated cysts that are more consistent in the medulla than in the cortex. The intervening renal parenchyma is usually normal, but those in older patients may have interstitial fibrosis, tubular atrophy, and glomerular sclerosis. Due to the variable cysts and parenchymal changes, some childhood ARPKD may histologically resemble ADPKD. The renal pelvis and ureter are normal . In the liver, the portal tracts characteristically exhibit bile duct proliferation and irregular anastomosis associated with fibrosis.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


