Fig. 11.1
Wilms tumor
Histologically, WT most commonly shows three distinct components that consist of blastemal, epithelial, and stromal elements (Fig. 11.2a). Biphasic and monophasic patterns may also occur. The epithelial component is usually the most recognizable element in WT and can show a variety of differentiation patterns. Epithelial differentiation into tubular structures is the most common differentiation pattern and ranges from rudimentary, rosette-like tubular structures to mature tubules. Primitive glomeruli may also be seen (Fig. 11.2b). Heterologous differentiation may occur and include mucinous or squamous epithelial, or neural, and neuroendocrine differentiation. The blastemal component shows either a sheetlike or nested architectural pattern and typically consists of small crowded blue cells with minimal cytoplasm, overlapping oval nuclei, and coarse chromatin (Fig. 11.2c). Mitoses are frequently seen. In the setting of blastemal-predominant WT, the differential diagnosis includes other pediatric small round blue-cell tumors such as neuroblastoma, rhabdomyosarcoma, primitive neuroectodermal tumor (PNET) , and lymphoma. Preoperative chemotherapy has been associated with the presence of necrosis, fibrosis, and macrophage influx, as well as formation of mature skeletal muscle. The stromal component varies from lightly staining spindle cells occasionally to rhabdomyoblasts and cartilage or bone [18–20].
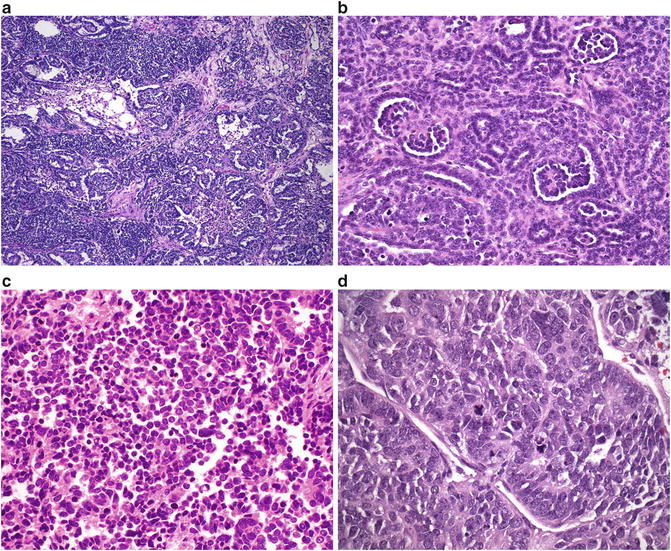
Fig. 11.2
(a) Wilms tumor: blastemal, epithelial, and stromal elements. (b) Wilms tumor: primitive glomerular structures. (c) Wilms tumor: blastemal component. (d) Wilms tumor showing anaplasia with hyperchromatic and enlarged nuclei and mitotic figures
Anaplasia occurs in approximately 5 % of WT and may be seen in the epithelial, stromal, or blastemal elements. It is more common in older children, reaching a peak at approximately 5 years of age [20]. Anaplasia is defined as hyperchromatic, markedly enlarged nuclei, and the presence of multipolar or polypoid mitotic figures (Fig. 11.2d). Focal anaplasia is defined as one or a few limited foci of anaplastic cells within a WT, with these anaplastic regions surrounded by areas without anaplasia or nuclear unrest approaching anaplasia. In contrast, diffuse anaplasia denotes a lesion that otherwise does not fit the “focal” definition and is associated with more aggressive behavior [21, 22].
Immunohistochemical analysis shows diffuse nuclear expression of WT1 in blastemal and epithelial cells, but not in stromal cells. In cases in which the stromal component is predominant, WT1 immunostain may not be a reliable indicator of the diagnosis [11, 23]. Epithelial elements can show cytokeratin immunoreactivity and blastemal cells may be positive for CD56 and negative for actin, myogenin, and MYOD1 [24–26].
Various genetic alterations may be associated with WT. These include deletion of the WT1 gene at 11p13, alterations of the WT2 gene at 11p15, p53 mutations, CTNNB1 gene mutations, and WTX mutations [27].
Prognosis and Clinical Management
Management of WT has been advanced primarily by two large cooperative multinational trials: (1) the National Wilms Tumor Study Group (NWTSG) , merged with three other groups to form the Children’s Oncology Group (COG), and (2) the International Society of Pediatric Oncology (SIOP) . SIOP and NWTSG differ in regard to risk stratification according to pathology. In SIOP, histological classification is divided into low-, intermediate-, and high-risk categories. Intermediate-risk tumors comprise 90 % of tumors and include epithelial-type WT, stromal-type WT, mixed-type WT, regressive-type WT and those with focal anaplasia. High-risk tumors, on the other hand, include blastemal-type WT and tumors with diffuse anaplasia. In NWTSG, histological classification is divided into two groups: favorable and unfavorable histology. Tumors with favorable histology include triphasic WT, epithelial-predominant WT, stromal-predominant WT, and blastemal-predominant WT. Tumors with unfavorable histology account for about 10 % of Wilms tumor and include tumors with either focal or diffuse anaplasia [15].
In SIOP and NWTSG, treatment protocols are based on stage and histology and involve various combinations of surgery, chemotherapy, and radiation. Whereas staging criteria are very similar between NWTSG and SIOP, there are fundamental differences between the two groups in regard to treatment of unilateral WT: primary surgery in NWTSG/COG versus initial or neoadjuvant chemotherapy in SIOP. In bilateral disease both NWTSG/COG and SIOP recommend preoperative chemotherapy. Radical nephrectomy is the mainstay of surgical therapy. Palpation of the renal vein before division is recommended to exclude the possibility of tumor thrombus. Intraoperative inspection of the liver and contralateral kidney is not required unless lesions were identified on preoperative imaging studies. Lymph node sampling is critically important, even in the setting of normal nodes on preoperative imaging. The role of partial nephrectomy has been suggested in cases of bilateral disease, as well as unilateral disease in the setting of syndromic conditions with increased risk of WT recurrence. Bilateral WT occurs in approximately 5 % of cases. Relapse occurs in about 15 % of children with favorable histology WT and 50 % of anaplastic histology WT. Most relapses occur early (within 2 years of diagnosis) and are found within the lungs (60 %) or abdomen (30 %). Currently survival is 90 % overall for children with Wilms tumor. Tumor histology and stage are the two most important prognostic factors [28, 29].
Premalignant Neoplasms Associated with Wilms Tumor: Nephrogenic Rests and Nephroblastomatosis
Introduction
Nephrogenic rests are clusters of residual embryonic metanephric cells, which normally disappear after 36 weeks of gestational age. The natural history of nephrogenic rests varies. Some regress and become sclerotic, while others have a hyperplastic overgrowth and may become neoplastic [30–32]. Nephrogenic rests can occur at any age but are most frequently found in infants [33]. It is estimated that they are present in about 1 % of unselected infants on postmortem biopsy [30]. Nephrogenic rests are classically divided into perilobar and intralobar types. Perilobar rests are much more common. They are usually found in subcapsular peripheral locations, are well demarcated, and often multiple. Intralobar rests, on the other hand, may be present anywhere within the kidney, often have a more irregular and intermixed margin, and are usually singular. Nephroblastomatosis is the presence of multiple or diffuse nephrogenic rests, and it can be perilobar or intralobar. Whereas perilobar nephroblastomatosis is found in association with synchronous bilateral WT, intralobar nephroblastomatosis is found in association with metachronous contralateral WT [33, 34].
Clinical Presentation
Nephroblastomatosis (NB) is generally asymptomatic. It refers to the presence of multiple or diffuse nephrogenic rests. The clinical significance of nephrogenic rests is their association with WT. They are found in about 40 % of kidneys with unilateral WTs and almost 100 % of kidneys with bilateral WTs [30]. Its presence in the non-tumoral part of kidneys with WT may also increase the risk of subsequent relapse of WT after treatment [35]. Overall, less than 1 % of nephrogenic rests will progress to WT [32]. Both nephrogenic rests and NB have been associated with multiple syndromes, including Beckwith-Wiedemann syndrome, hemihypertrophy, Perlman syndrome, and trisomy 18 [30, 35].
On ultrasound, NB appears isoechoic or hypoechoic compared to the cortex, with small lesions being easily missed. On CT scan, the lesions are homogeneous and isodense on pre-contrast imaging, then become hypodense compared to the cortex on post-contrast imaging. Occasionally, regions of NB are so small they can be missed with CT as well [36]. On MRI, NB is isointense or slightly hypointense to the cortex on both T1-weighted and T2-weighted imaging. On gadolinium-enhanced T1-weighted images, the lesions remain homogeneously hypointense compared with the enhanced cortex. The major difference with NB and WT is the fact that the latter tends to show mixed echogenicity and heterogeneous structures on imaging [37]. An open biopsy that includes a portion of the lesion plus adjacent normal renal cortex can be helpful in distinguishing NB from WT. In WT, a pseudocapsule consisting of compressed renal parenchyma surrounds the tumor, while such a capsule does not exist with NB.
Pathology
Perilobar rests are often multifocal and occur along the periphery of a renal lobe. The foci are well circumscribed and contain both blastemal and tubular elements with only scant stroma (Fig. 11.3a). In contrast, intralobar rests are randomly situated within the renal parenchyma, are poorly circumscribed, and show a prominent stromal component (Fig. 11.3b). Both subtypes can be further subdivided into sclerosing (fibrotic background), obsolete (predominant fibrosis), dormant (small foci), and hyperplastic (large foci with abundant mitosis) subtypes. The distinction between hyperplastic RN and WT can be difficult, but may be more readily made if the border of the lesion is well sampled and surveyed for the presence of a pseudocapsule [30, 32].
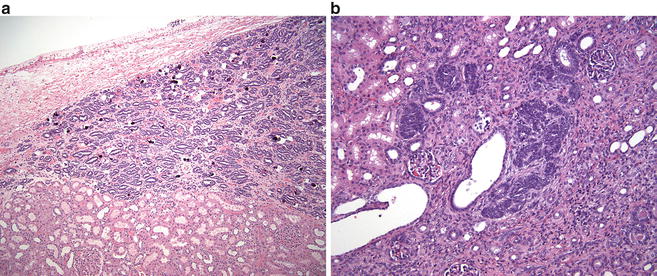
Fig. 11.3
(a) Perilobar nephrogenic rests contain both blastemal and tubular elements with scant stroma. (b) Intralobar nephrogenic rests with prominent stromal component
Nephroblastomatosis is characterized by the presence of multifocal and diffuse nephrogenic rests lesions. Nephroblastomatosis can be subclassified as perilobar, intralobar, combined, or panlobar and can display all or any subclasses of nephrogenic rests patterns (dormant, hyperplastic, and neoplastic). Architecturally, nephroblastomatosis may be associated with a thick cortical outer layer of hyperplastic nephrogenic tissue [37, 38].
Similar to WT, NRs may contain alterations in the WT1 and WT2 genes. One model for the development of WT is the accumulation of one or more additional mutations within the NRs.
Prognosis and Clinical Management
The treatment of NB remains controversial [30, 35]. It is a benign process, yet has a malignant potential [39]. Neither the National Wilms Tumor Study Group (NWTSG) nor the International Society of Pediatric Oncology (SIOP) provides formal guidance on how to treat NB or NRs in the absence of WT. As a consequence, patients with NB are treated in various ways, some with multiple chemotherapeutic regimens and some not at all [40]. The arguments in favor of chemotherapy for NB are (1) to treat undiagnosed WT within the NB lesion, (2) to decrease the number of target cells at risk for malignant transformation, and (3) to prevent damage to the normal renal cortex by compression from massive NB [30–32, 35]. The arguments against initial chemotherapy are (1) the risk of chemotherapy for treatment of a benign disease that may regress spontaneously, (2) the uncertainty that chemotherapy will prevent the development of WT, (3) the fear that chemotherapy will select for chemoresistant cells, and (4) the risk of NB regrowth after chemotherapy [38, 41]. There are sporadic case reports of spontaneous resolution of NB without treatment; however, the risk of developing WT persists even years after initial diagnosis.
Clear Cell Sarcoma of the Kidney (CCSK)
Introduction
Clear cell sarcoma of the kidney (CCSK) comprises approximately 5 % of all renal tumors in children. It is the second most common pediatric renal tumor after Wilms tumor [42–44]. The tumor presents at a mean age of about 36 months and rarely occurs in the first 6 months of life [44–59]. Recently a t(10;17)(q22;p13) translocation has been identified in CCSK, suggesting its possible role in tumorigenesis [60–62]. Gene expression profiles of CCSK suggest the cell of origin to be a renal mesenchymal cell with neural markers.
Clinical Presentation
Presenting symptoms are similar to that of other pediatric renal tumors and include abdominal mass, abdominal and flank pain, and hematuria. Distributions by stages are 27 % for stage I, 33 % for stage II, 34 % for stage III, and 6 % for stage IV [42, 44, 45, 62–67]. Bilateral (stage V disease) has only been reported in a few case reports [44, 68, 69]. The most common sites of metastases are lymph nodes (60 %) and bone (13 %), from which it derives the name “bone metastasizing renal tumor of childhood.” Other metastatic sites include the brain, lung, liver, skin, and colon. CCSK is also able to invade the vena cava with extension into the right atrium. No specific syndromic or familial associations have been established [44, 45, 52, 66, 69, 70].
Pathology
CCSK is typically a unifocal, well-circumscribed renal mass that is often large (mean diameter 11.3 cm). The cut surface shows a solid, homogeneous tan-gray appearance (Fig. 11.4a). Cystic structures, focal necrosis, and hemorrhage may be present [44].
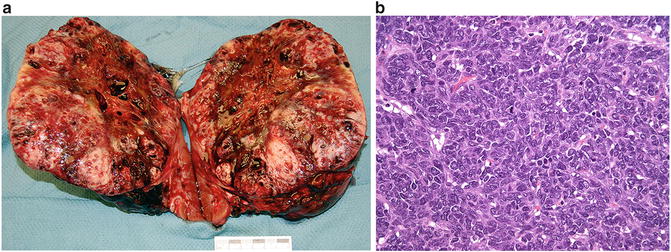
Fig. 11.4
(a) Clear cell sarcoma of the kidney. (b) Clear cell sarcoma of the kidney showing dense populations of monotonous appearing cells
Microscopically, CCSK shows a densely cellular proliferation of monotonous appearing cells with clear to mildly eosinophilic cytoplasm (Fig. 11.4b). Nuclei are round to oval, contain fine granular chromatin, and lack prominent nucleoli. Mitotic figures are frequent and may be atypical. Tumor cells invade into the adjacent renal parenchyma. The lesional background shows delicate branching vessels and occasional necrosis, which is an adverse prognostic indicator. Mucopolysaccharides may be present in the surrounding matrix. Several variants of CCSK have been described and include myxoid, sclerosing, epithelioid trabecular, palisading (Verocay body-like), spindle cell, and anaplastic forms [44, 48, 52, 71–73].
Prognosis and Clinical Management
Current treatment strategies involve various combinations of surgery, chemotherapy, and radiation depending on stage. Although the optimal treatment for CCSK has not yet been established, with current intensive treatment schedules, 5-year relapse-free survival (RFS) now ranges from 75 to 85 % and 5-year overall survival (OS) from 85 to 90 % [44, 67, 73]. Younger children tend to have poorer outcomes (i.e., lower survival and higher recurrence rates) [67]. Relapses occur in 20–40 % of cases. Time interval to relapse is u sually between 12 and 24 months, although late relapse up to 8 years after treatment has been reported [42, 44, 48, 52, 55, 64, 66, 67, 72, 77–81]. The most recent studies indicate that the brain has now surpassed bone as the most common site of CCSK recurrence [67, 77].
Rhabdoid Tumor of the Kidney
Introduction
Rhabdoid tumor of the kidney (RTK) is a highly aggressive malignant neoplasm, characterized by early metastases and a high mortality rate. It occurs primarily in the infant and newborn and is the second most common malignant tumor in the neonate [82]. Eighty percent of RTKs occur in patients under the age of 2 years and 60 % occur in patients under the age of 1 year [83]. After the age of 5 years, the diagnosis is unlikely [82–89]. The histologic origin of RTK remains obscure. RTK was named because microscopically it resembles rhabdomyosarcoma , although it does not show skeletal muscle markers either by staining or electron microscopy [18, 88].
Clinical Presentation
The clinical presentation of children with RTK has not been well described. Gross hematuria can be seen in greater than 50 % of children with RTK and microscopic hematuria can be seen in nearly 75 % of patients. Hematuria as a presenting symptom suggests invasion of the renal pelvis by tumor [90–92]. RTK can also been diagnosed in utero by ultrasound and may present in the newborn as metastases to the skin or other sites [84, 86–88, 92–94]. Sites of metastasis include regional lymph nodes, the lungs, the liver, bone, and the brain [95]. Hypercalcemia has also been associated with RTK and is attributed to an increased serum PTH [96, 97]. Radiographically, RTK is indistinguishable from other malignant pediatric renal tumors. On CT, a prominent and eccentric crescent with attenuation of fluid—representing subcapsular renal hemorrhage or peripheral tumor necrosis adjacent to tumor lobules—may be seen in up to 71 % of cases; however, this finding is not specific to RTK [83].
Pathology
On gross evaluation, RTK is a large, unencapsulated tumor that often overruns the kidney and extends into the perirenal tissue. The cut surface is often necrotic and hemorrhagic (Fig. 11.5a). Tumors can range up to 17 cm in size (mean 9.6 cm) [18, 82, 83, 86].
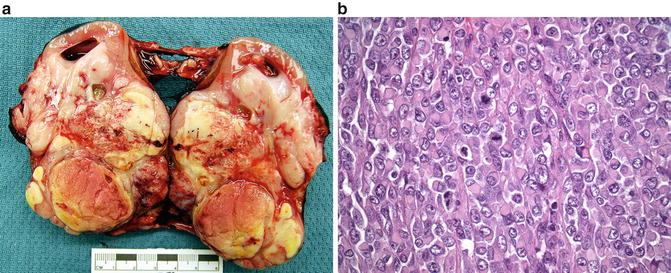
Fig. 11.5
(a) Rhabdoid tumor of the kidney. (b) Rhabdoid tumor of the kidney: large polygonal cells with dense eosinophilic cytoplasmic inclusions and mitotic figures
Histological evaluation shows sheets of loosely cohesive cells that extensively invade the surrounding renal parenchyma and perirenal tissue. Tumor cells are large and polygonal, often showing dense eosinophilic cytoplasmic inclusions that likely represent aggregates of intermediate filaments (Fig. 11.5b). Large, atypical vesicular nuclei are readily apparent, which contain clumped chromatin and prominent nucleoli [87–89, 98–101].
Prognosis and Clinical Management
Surgical excision is the mainstay of treatment, followed by adjuvant chemotherapy and/or radiotherapy. Prognosis associated with RTK is generally poor despite multimodal therapy [102].
Desmoplastic Small round Cell Tumor of the Kidney
Introduction
Desmoplastic small round cell tumor (DSRCT) is a rare, high-grade, malignant neoplasm usually seen in children, adolescents, and young adults, where it has an aggressive clinical course [103–107]. Recently a few cases involving the kidney have been reported in children (age 6–8 years of age) [108]. DSRCT is associated with a specific chromosomal translocation, t(11;22)(p13;q12), the identification of which is a requisite for diagnosis . The t(11;22)(p13;q12) translocation product is a chimeric transcript formed by fusion of the Ewing sarcoma gene (EWS) and the Wilms tumor gene (WT1), thought to be responsible for tumorigenesis [109].
Clinical Presentation
Most of these tumors manifest as multinodular masses associated with the serosa of the abdominal cavity and usually minimal organ involvement. When these tumors involve the urogenital system, they are generally metastatic at the time of presentation and tend to have a poor prognosis [105, 110]. DSRCT specific to the kidney may present as an abdominal mass with associated symptoms that can include cramping, abdominal pain, weight loss, and constipation. It has been suggested that on imaging, these tumors may appear as hypovascular, well-circumscribed renal masses with associated punctate calcifications; however, the lack of these characteristics does not exclude the diagnosis [111].
Pathology
On gross evaluation, DSRCT is often a solitary white-tan, nonencapsulated mass that can invade into the surrounding renal parenchyma and perirenal tissue. Necrosis and hemorrhage are common and tumor can range up to 13 cm in size [108, 112–114]. On microscopic assessment, nests of undifferentiated cells can be seen, occasionally associated with necrosis and calcification. The tumor cells display scant cytoplasm, and the nuclei are large and hyperchromatic. Mitotic figures are frequent. Very few pediatric cases of DSRCT have been reported in the literature; most describe an absence of desmoplasia, which makes DSRCT indistinguishable from other small round cell tumors [105, 108, 112, 114–117]. Molecular studies are essential for the diagnosis by identifying t(11;22)(p13;q12) translocation. Additionally, immunoreactivity for vimentin, desmin, cytokeratin, EMA, and WT-1 can also be seen [108, 111, 113].
Anaplastic Sarcoma of the Kidney
Introduction
Clinical Presentation
The most common presentation is a palpable abdominal mass and sometimes hematuria. Anaplastic Wilms tumor is the most important tumor in the differential [122].
Pathology
On gross examination, ASK is a large, infiltrative lesion that shows frequent cystic change. Size ranges up to 21 cm (mean 12.7 cm) [122]. Histological review shows bundles of undifferentiated spindle cells that merge with small round primitive mesenchymal cells. The tumor background often shows myxoid change. Anaplastic foci that may demonstrate giant pleomorphic cells with enlarged irregular hyperchromatic nuclei can be seen throughout the tumor. Prominent areas of chondroid differentiation may be present, as well as small regions that contain osteoid with osteoclast-like giant cells [120–123].
Prognosis and Clinical Management
Considering patients have been treated with various therapeutic protocols (nephrectomy, chemotherapy, and/or radiation), the overall outcome of ASK is reasonably good, with 75 % of patients in one series surviving after 8 years of follow-up [122].
Indolent Renal Lesions and Malformations
Congenital Mesoblastic Nephroma (CMN)
Introduction
Congenital mesoblastic nephroma (CMN) is the most common renal tumor of young patients up to 3 months of age and accounts for 2–3 % of all pediatric renal tumors [124]. The median age at diagnosis is 30 days, with 90 % occurring within the first year of life and virtually none after the age of 3 years [125, 126]. Histologically, three variants are recognized. The classic type accounts for about 24 % of cases, the cellular type accounts for about 66 % of cases, and a mixed variant of both classic and cellular types comprises 10 % of cases. The cellular variant CMN is specific for a t(12;15)(p13;q25) translocation with resultant ETV6-NTRK3 gene fusion transcript [127, 128]. Interestingly, this same translocation is shared by infantile fibrosarcoma (IFS), suggesting that cellular CMN represents intrarenal IFS [129].
Clinical Presentation
CMN is usually detected prenatally by ultrasonography. In the infant, it most commonly presents as a palpable abdominal mass. On imaging, CMN is predominantly solid, but cystic or even calcified components may be identified [130]. Though CMN is largely a benign tumor, cases of metastases, especially of the cellular variant, have been reported. Reported sites of metastases include the brain, lung, liver, and heart.
Pathology
On gross examination, CMN is often a unifocal mass that may replace a large portion of the kidney (Fig. 11.6a). The cut surface is gray to tan in color and shows a trabeculated, firm appearance. Cystic change, necrosis, and hemorrhage are occasionally present. Microscopically, CMN is comprised of fascicles of spindled cells with minimal atypia that often intercalate between adjacent renal elements (Fig. 11.6b). Mitotic figures are rare. A cellular variant of CMN has been described, which shows an increase in cellularity associated with a high mitotic rate, raising the differential diagnosis of infantile fibrosarcoma (Fig. 11.6c) [125, 126, 131, 132]. Both CMN and infantile fibrosarcoma have been reported to contain the t(12;15)(p13;q25) translocation, suggesting they may be potentially related entities [133].
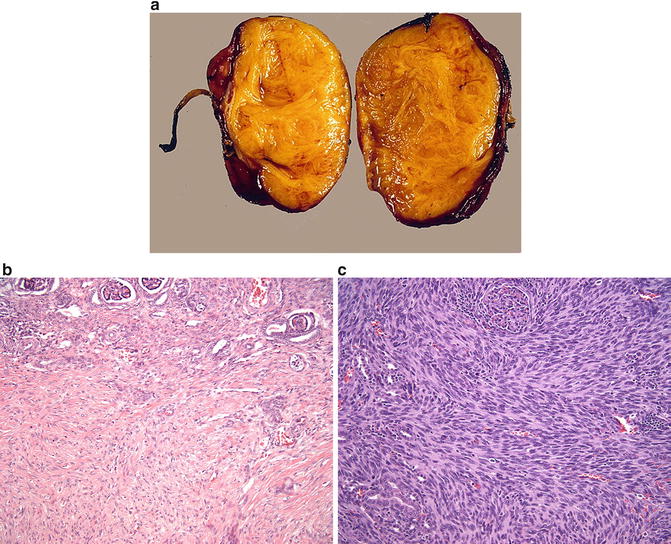
Fig. 11.6
(a) Congenital mesoblastic nephroma. (b) Congenital mesoblastic nephroma, conventional pattern. (c) Congenital mesoblastic nephroma, cellular variant
Prognosis and Clinical Management
Complete surgical resection remains the most important prognostic criterion [131, 135]. Radical nephrectomy is preferable to partial nephrectomy in order to reduce the risk of local recurrence. Overall survival is greater than 95 % with a worse prognosis associated with older age, stage III disease, and the cellular variant [131]. Relapse occurs in only 5 % of patients and is thought to be related to incomplete resection. Recurrences almost always develop within 12 months of surgery; thus, close surveillance is recommended during this period of time [136]. Cases that recur locally have been successfully treated with chemotherapy, with or without radiotherapy, with good outcomes [137].
Metanephric Stromal Tumor
Introduction
Metanephric stromal tumor (MST) is a benign pediatric tumor, most commonly seen in the first decade of life (mean age 2 years old). It is a rare renal pediatric neoplasm that is equally seen in both genders. MST was first described by Argani and Beckwith and was previously grouped as congenital mesoblastic nephroma (CMN). The distinction was clear given MST’s benign behavior , which lacks the degree of microscopic infiltration seen in CMN [138–143].
Clinical Presentation
Clinically, MST is often asymptomatic and presents itself as an incidental abdominal mass. Other symptoms, such as hypertension with hyperreninism, are seen when juxtaglomerular apparatus hyperplasia is involved and hematuria when the renal pelvis is involved. Radiologic findings are inconclusive, and diagnosis is made through pathologic analysis of excised tumor [138, 140, 142, 143].
Pathology
MST is most commonly a solitary lesion in the renal medulla that shows a lobulated, tan to yellow appearance on cut surface. Small cysts may occasionally be present. Most MSTs are small to medium lesions, with an average approximate size of 4 cm [140]. On microscopy, bland stromal cells surround blood vessels and background renal parenchymal structures, giving an appearance of concentric rings (Fig. 11.7). A myxoid background may be present. Angiodysplasia may be present and is characterized by vessels showing epithelioid transformation of the medial smooth muscle. Heterologous elements including glial tissue or cartilage may be present [140, 144].
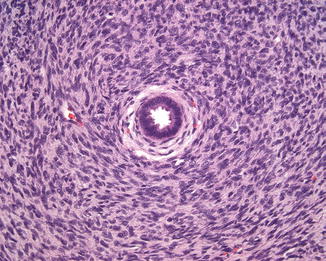
Fig. 11.7
Metanephric stromal tumor
The differential diagnosis of MST includes CMN and CCSK. In contrast to the deeply interdigitating nature of CMN into the background renal parenchyma, MST is typically smaller with more focal regions of intercalation into the surrounding kidney. MST is occasionally more difficult to distinguish at the cytological level from CCSK, although the more limited nature of MST lesions and the absence of the branching vascular pattern seen in CCSK can help in the distinction of these two entities. Immunohistochemical stains in MST show focal expression of CD34 (which is negative in CMN and CCSK) and absence of desmin, cytokeratin, and S100 immunoreactivity [134, 138, 140–144].
Metanephric Adenofibroma
Introduction
Metanephric adenofibroma (MAF), also called nephrogenic adenofibroma , is a rare biphasic tumor that combines the epithelial component of metanephric adenoma (MA) and stromal component of MST. Few cases have been reported in the literature with age ranging from 5 months to 36 years old (mean 6 years). It is more commonly seen in the male population (2:1). MAF can be morphologically similar to Wilms tumors (WT) and intralobar nephrogenic rests, and it has been considered a hyperdifferentiated version of these tumors [134, 144, 145].
Clinical Presentation
Pathology
MAF is localized to the renal medulla and is most commonly seen as a solitary lesion with a tan to yellow cut surface. Cystic change and nodularity have been reported. Average gross size is 3.85 cm [145]. Microscopically, there are a variable proportion of epithelial and stromal elements in MAF. The stromal element appears identical to that seen in MST, including the “onion skin” patterning, although is less likely to be associated with juxtaglomerular hyperplasia. The epithelial element shows unencapsulated groups of epithelial cells separated by bands of stroma. Numerous variants have been reported that overlap in appearance with more conventional metanephric adenoma. Immunohistochemical features of the stromal cells are similar to MST, with immunoreactivity to CD34 and absence of staining for desmin, muscle-specific actin, and cytokeratin. The epithelial component, however, is often immunoreactive for keratins and negative for AMACR [134, 144, 145].
Multicystic Dysplastic Kidney
Introduction
Multicystic dysplastic kidney (MCDK) is a nonneoplastic lesion characterized by malformation of the kidney that manifests as variably sized cysts and frequently absence or diminishment of the vascular pedicle and ureter. Cysts may enlarge, remain stable in size, or involute [149–154]. The etiology of MCDKs remains unknown, but there are two leading theories. The obstruction theory proposes that the MCDK results from severe fetal obstructive hydronephrosis due to atresia of the renal pelvis or ureter [155]. The other theory suggests perturbation of the interaction between the ureteric bud and metanephric blastema [156, 157]. MCDK usually develops as a sporadic problem, although familial occurrence has been reported. The reported incidence of MCDK is estimated between 1 in 1000 and 1 in 4300 live births [158–161].
Vesicoureteral reflux (VUR) , ureteropelvic junction (UPJ) obstruction, and megaureter of the contralateral kidney occur in children with MCDK more frequently than in the general population [152, 161–170]. VUR is the most frequent finding, with a reported incidence of up to 43 % in patients with MCDK. Therefore, VCUG is often recommended early in life. The majority of cases of VUR in these patients are asymptomatic, low grade, and self-limiting [152, 161–171]. The risk of hypertension among children with MCDK is similar to that seen in the general pediatric population [172–175]. While hypertension has been reported in association with retained MCDKs, it is likely a result of damage to the contralateral functional solitary kidney rather than the MCDK itself [152, 159, 176]. Hypertension associated with MCDK may or may not resolve after nephrectomy of the MCDK [177–179]. There is a slight increased risk of Wilms tumor in patients with MCDK, with an incidence of 1 in 10,000 in the general population compared to 3 to 10 in 10,000 in children with MCDK in the USA. Approximately 16,000 MCDKs would have to be removed to prevent one Wilms tumor-related death and neither surveillance nor prophylactic nephrectomy seems warranted [180–184].
Clinical Presentation
Most cases of MCDK are diagnosed on prenatal ultrasonography and less often because of a palpable abdominal mass. Most patients have neither symptoms nor a palpable abdominal mass [152]. The majority of cases of MCDK undergo spontaneous involution. Great variation in the prevalence of involution has been reported, ranging from 10 % at a mean of 33.5 months to 75 % at a mean of 16 months of observation [167, 179, 185–188]. Initially smaller MCDKs are more likely to involute and do so at an earlier age [189]. The contralateral kidney is often hypertrophic with compensatory growth.
Pathology
On gross examination, the kidney may be diffusely involved by variably sized cysts and the pelvicalyceal system and ureter may be narrow and misshapen. On occasion, the process may be bilateral. On microscopy, immaturity and disorganization of the renal parenchyma are apparent. Epithelial structures are commonly present and include primitive ducts lined by cuboidal to columnar epithelium, cysts, and rudimentary glomerular elements (Fig. 11.8). Fibrous stroma may be present and can include the presence of heterologous cartilage [157].
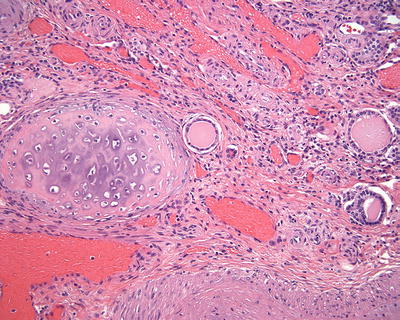
Fig. 11.8
Multicystic dysplastic kidney
Prognosis and Clinical Management
Management involves observation and treatment of associated conditions. Indications for nephrectomy are restricted to large MCDKs that interfere with respiratory or intestinal function in the neonatal phase and those that contain solid components that grow on serial imaging [152]. Prognosis depends on whether involvement is unilateral or bilateral and on the presence and severity of associated anomalies. Bilaterality results in increased fetal loss or profound oligohydramnios with rapid respiratory death after birth [156]. Most patients with isolated unilateral MCDK, however, do not experience any problems or complications as a consequence of this congenital anomaly.
Medullary Sponge Kidney
Introduction
Medullary sponge kidney (MSK) results from a disruption of the ureteric bud-metanephric mesenchyme interface due to abnormalities in the glial cell line-derived neurotrophic factor (GDNF) or receptor tyrosine kinase (RET) genes [190, 191]. Defective acidification is the initial abnormality, which is followed by a sequence made up of defective bone mineralization, hypercalciuria, and stone formation. Patients may be in a negative calcium balance, which can cause hyperparathyroidism . Defective urinary concentrating ability and distal tubular acidosis can also be found which is a result of the functional abnormality of the terminal collecting ducts [192]. Exact prevalence is unknown although it is probably less than 0.5–1 %. It is much more frequent (12–20 %) in recurrent renal calcium stone formers [193].
Clinical Presentation
MSK is generally diagnosed in young adulthood. Three different clinical profiles are recognized, all of which show the typical radiological feature of ectatic precalyceal papillary collecting ducts and nephrocalcinosis. The two most common include an indolent form rarely characterized by stones and diagnosed incidentally or with atypical signs and a much more prevalent form in which the patients manifest with many small stones that occasionally need urologic intervention. The third profile is a rare but severe condition dominated by intractable excruciating renal pain [194]. Urinary tract infection is considered to be the second most frequent clinical problem after renal stones in patients with MSK. Pyelonephritis can occur from urine stagnation in the dilated prepapillary tubule or by stones themselves. Recurrent infections can cause struvite stones, which may rarely lead to ESRD. Occasionally patients also discuss passing of sand in their urine. Diagnosis historically has been done via intravenous urogram to visualize tubular ectasia. With the increase in the use of CT and ultrasound, diagnosis is sometimes made erroneously based on multiple small stones which may or may not have the associated ectasia, which is specific for MSK [195].
Pathology
On gross evaluation, the papillae in MSK are enlarged with rounded contours and blunted tips. The papillae may also show white and yellow plaques and variable numbers of stones. The classic histological triad for MSK consists of undifferentiated interstitial cells, multilayered hyperplastic epithelium of the medullary collecting ducts, and single-layered epithelium in dilated medullary collecting ducts. Small stones may be present in the latter structure. Immunohistochemical stains show expression of Runx2 in the nucleus of interstitial cells [193, 194, 196, 197].
Prognosis and Clinical Management
Treatment with potassium citrate aids with the observed hypercalciuria and hypocitraturia [193]. A diet with high fluid intake, low salt, and low protein is also recommended for overall stone prevention, and thiazide diuretics are also used as an adjunct. Surgical intervention can be necessary as a result of infected or obstructing stones. Endourological approaches are available as well as complex reconstructions including autotransplantation with creation of a pyelovesicostomy to allow for stone passage [198].
Reflux Nephropathy
Introduction
Reflux nephropathy has been used to describe the renal abnormalities in patients diagnosed with vesicoureteral reflux (VUR) . These lesions can include renal scarring, renal lesions, renal parenchymal abnormalities, dysplasia, and renal hypoplasia [199]. Several factors may contribute to reflux nephropathy including the severity of the VUR as well a possible genetic predisposition. VUR in itself is quite common with estimates at 1–2 % of all live births [200]. It is unknown how many of these children go on to have manifestations of reflux nephropathy. Familial clustering of VUR implies that genetic factors have an important role in its pathogenesis, but no single major locus or gene for VUR has yet been identified and most researchers now acknowledge that VUR is genetically heterogeneous [201]. Since VUR is relatively common, there are a multitude of associated conditions along the CAKUT (congenital abnormalities of the kidney and urinary tract) spectrum. Many syndromes such as renal coloboma, Kallman, urofacial, de Lange, Bardet-Biedl, and Epstein syndrome can also be seen. The inheritance in these syndromes is usually autosomal dominant and multiple genes have been found to be disrupted including PAX2 [199, 200, 202–205].
Clinical Presentation
In actuality, reflux nephropathy is a pathological entity that ideally would be documented histologically. However clinically it is diagnosed with recurrent urinary tract infections (UTIs) in the context of VUR and evidence of radiological abnormalities. A DMSA scan (dimercaptosuccinic acid renal scan) is the ideal imaging to confirm the presence of abnormalities although intravenous pyelography (IVP) has been used in the past. The differentiation between renal hypoplasia/dysplasia and reflux nephropathy can be difficult based on imaging alone, but the lack of UTIs is consistent with renal hypoplasia/dysplasia where the abnormality is in ureteral bud development.
Reflux nephropathy can cause long-term consequences of hypertension, loss of renal function, and even end-stage renal disease. Hypertension is thought to be mediated by the renin-angiotensin system with higher levels of serum renin. Eighteen percent of patients were noted to have hypertension after 15 years with initial findings of reflux nephropathy. Most patients become hypertensive between the ages of 15 and 30 [203]. In some studies hypertension can occur with any degree of renal damage [204]. Problems with hypertension can also manifest as preeclampsia during pregnancy. The incidence of preeclampsia was noted to be 24 % in women with bilateral renal scars versus 7 % with unilateral renal scarring [202]. The Italian Pediatric Registry of Renal Failure reported that 25 % of patients with end-stage renal failure had a history of VUR [206].
Pathology
Reflux nephropathy can be associated with inflammation and occasional colonization by bacterial organisms. The inflammatory response can cause local renal damage with deposition of collagen and destruction of the renal parenchyma over time. Histologically, the areas of damaged renal parenchyma show interstitial fibrosis, glomerulosclerosis, and dilated and atrophic tubules [199, 207].
Prognosis and Clinical Management
The treatment of VUR continues to be controversial. The use of prophylactic antibiotics and surgical management can vary among providers. Ideally VUR will resolve after a period of observation. If it doesn’t resolve, treatment can depend on the presence of scarring, the current grade of VUR, and parental or provider bias [208].
Hematologic Malignancies Involving the Pediatric Kidney
Renal Lymphoma and Leukemia
Introduction
Primary renal lymphoma and leukemia are exceedingly rare in children [209, 210]. More often, renal involvement results from infiltration of the kidney by circulating malignant cells. Once within the kidneys, tumor infiltration is initially interstitial, with the nephrons, collecting ducts, and blood vessels acting as scaffolding for tumor growth. Further growth leads to destruction of the renal parenchyma scaffolding, resulting in an expansile growth pattern. Since only a fraction of these patients undergo CT imaging, the true incidence of renal leukemic involvement in children is unknown [211].
Clinical Presentation
Renal lymphoma may present with renal failure, abdominal pain and abdominal mass, anemia, or hematuria [212–220]. On imaging, kidneys may appear unilaterally or bilaterally enlarged, as a single lesion, or as multiple mostly cortical lesions [221–223]. Regional lymph node involvement is common at presentation, as are metastases to the lungs, liver, choroid plexus, tonsils, and bone marrow [215, 220, 224–228]. Lymphoma infiltration may affect the kidney either primarily or secondarily [229, 230]. Secondary renal involvement as a part of systemic disease or contiguous infiltration from adjacent lymphoma is detected in only 3–8 % of all patients, most commonly through routine CT staging for lymphoma [222, 231–234]. In autopsy series, however, estimates of renal involvement in patients with known lymphoma range from 30 to 60 % [235].
Nephromegaly, hematuria, and renal dysfunction may be seen [209, 210, 224, 236–239]. However, renal leukemic involvement is usually an incidental finding. Unlike patients with lymphoma, children with leukemia do not generally require routine CT imaging for staging or follow-up. Instead, CT imaging in children with leukemia is typically utilized in the assessment of possible disease-related complications or for the evaluation of some other clinical problems. Renal leukemic involvement can present with a wide variety of contrast-enhanced CT imaging findings. The most common focal parenchymal abnormality is that of multiple bilateral renal low-attenuation masses. These may appear as small and large round low-attenuation masses, wedge-shaped and geographic low-attenuation masses, or ill-defined areas of low attenuation. Diagnosis involves core needle biopsy in association with hematologic findings [211].
Pathology
Lymphomas and leukemias involving the kidney may appear as either a focal lesion or as diffuse involvement of the kidney on gross examination. The cut surface appears firm and pale, often with a homogeneous appearance. Necrosis, hemorrhage, cystic change, calcifications, and tumor thrombi can occur. Microscopically, abnormal lymphoid cells are present infiltrating between background renal elements [134]. These lymphoid cells may be present in sheets, which is often associated with diffuse renal enlargement, or as discrete nodules. Malignant lymphoid cells may also be primarily present within vascular spaces (intravascular lymphoma).
Immunohistochemical, flow, and molecular analyses can help to identify the lymphoid lineage of the neoplastic cells [134, 209, 211, 239].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


