Syndrome (abbreviation)
Gene (protein)
Chromosome location
Renal pathology
Other clinical manifestations
Von Hippel-Lindau (VHL) syndrome
VHL (pVHL)
3p25
Clear cell RCC, cystic renal cell carcinoma, renal cysts
Central nervous system hemangioblastomas, pheochromocytomas, pancreatic cysts, neuroendocrine tumors; endolymphatic sac tumors; epididymal and broad ligament cystadenomas
Hereditary papillary RCC (HPRCC)
MET (HGFR)
7q31
Papillary type 1 RCC
None
Hereditary leiomyomatosis and RCC (HLRCC)
FH (pFH)
1q42-43
Papillary type 2 RCC; often solitary
Uterine leiomyomas and leiomyosarcoma, cutaneous leiomyomas
Birt-Hogg-Dubé (BHD) syndrome
FLCN (Folliculin)
17p11.2
Hybrid oncocytic RCC, chromophobe RCC, clear cell RCC, oncocytoma, oncocytosis
Cutaneous papules, lung cysts, spontaneous pneumothoraces
Tuberous sclerosis (TS) syndrome
TSC1 (hamartin)
9q34
Angiomyolipoma, papillary RCC, hybrid oncocytic RCC
Facial angiofibromas, periungual fibromas, shagreen patches, cardiac rhabdomyomas, lung and kidney cysts, cortical tubers
TSC2 (tuberin)
16p13
Succinate dehydrogenase mutation
SDHB (pSDHB)
1p36
B – solid and cystic tumors, variable pathology, oncocytic and clear cell RCC
Paragangliomas/pheochromocytomas, GIST, pituitary adenomas, pulmonary chondromas
SHDC (pSDHC)
1q23
C – solid tumors, clear cell RCC, low grade
SDHD (pSDHD)
11q23
D – solid tumors, clear cell RCC
Cowden/PTEN hamartoma tumor syndrome
PTEN (pPTEN)
10q23
Variable pathology, including clear cell, chromophobe, and papillary RCC
Breast, thyroid, and endometrial cancer, mucocutaneous lesions, acral keratoses, facial trichilemmomas, papillomatous papules, macrocephaly, Lhermitte-Duclos disease, benign thyroid conditions, mental retardation, GI hamartomas, lipomas, fibromas, breast fibrocystic disease
Microphthalmia transcription (MiT) family translocations
Fusion chimeric proteins with TFE3 or TFEB
Xp11.2
Solid or cystic tumors, variable architecture RCC
None
t(6;11)
Microphthalmia-associated factor mutation
MITF (pE318K)
3p14
Solid or cystic tumors, clear cell and papillary RCC
Melanoma
Chromosome 3 translocation
Fusion chimeric protein
t(3;8)
Clear cell RCC
None
BRCA1-associated protein-1 (BAP1) mutation
BAP1 (pBAP1)
3p21
Solid and cystic tumors, high-grade clear cell RCC
Uveal/cutaneous melanoma, mesothelioma
Von Hippel-Lindau
Introduction
Von Hippel-Lindau (VHL) syndrome is a hereditary disease with an autosomal dominant pattern . The VHL incidence is estimated to about 1:36,000 [7]. Patients usually develop characteristic neoplasms in multiple organs, including clear cell RCC, capillary hemangioblastomas of the central nervous system and retina, pheochromocytoma, pancreatic tumors, and inner ear tumors [7]. The mean age of RCC onset in patients with VHL syndrome is 37 years, which is much earlier than that for sporadic RCC (61 years).
VHL disease is caused by germline mutations of the VHL gene, which is located on the short arm of chromosome 3 (3p26-25) [8]. The VHL gene is a relatively small tumor suppressor gene with a coding sequence of 639 nucleotides on three exons [9]. The VHL protein is responsible for proteomic degradation of hypoxia-inducible factors (HIFs) [10]. HIF-1α and HIF-2α regulate a number of downstream genes, including vascular endothelial and platelet-derived growth factors (VEGF and PDGF), transforming growth factor-α (TGF-α), erythropoietin, glucose transporter protein-1, carbonic anhydrase IX, and C-mesenchymal-epithelial transition factor (c-MET) [2]. These downstream genes are involved in the cell cycle, angiogenesis, and tumorigenesis. Mutations of the VHL gene, which may occur in any exon, are most commonly missense mutations, but nonsense mutations such as deletions, insertions, and translocations may also occur. The germline mutation is found in nearly all VHL families [11]. The type of mutation is associated with the clinical presentation of VHL disease. Somatic VHL alterations are also common in sporadic clear cell RCCs, but they are usually absent in papillary, chromophobe, or collecting duct RCCs.
Clinical Presentation
The earliest manifestations of VHL are usually not related to renal pathology. Central nervous system (CNS) hemangioblastomas, including retinal hemangioblastomas, can present as vision loss or neurologic symptoms as early as age 1 [7]. Other early manifestations in specific families include pheochromocytoma and pancreatic cysts, with ages of onset as young as 5 having been reported. Renal tumors have been noted as early as the teenage years, but generally present in the 4th decade of life on routine screening exams or with gross or microscopic hematuria. Overall some 25–60 % of VHL patients develop renal tumors, renal cysts, or cystic renal cell carcinomas; these are often bilateral and multifocal, as with most all forms of FRCC. Therefore, screening efforts for renal tumors in individuals affected by VHL generally start at age 18, when annual axial imaging (CT or MRI) is recommended .
Pathology
Grossly, the tumors often present as multiple, well-circumscribed, bright yellow masses in both kidneys. There are often numerous cysts containing clear fluid in the kidneys. Some patients may develop up to 600 microscopic tumors and over 1000 cysts in each kidney [12]. Microscopically, the tumors are typically composed of clear cell RCC, which is characterized by nests or sheets of tumor cells with clear cytoplasm separated by a delicate vascular network (Fig. 6.1a). The tumor may show focal papillary, tubular, or cystic features (Fig. 6.1b). The renal cysts are usually lined by a single layer of tumor cells with clear cytoplasm, sometimes forming small tufts and papillary structures (Fig. 6.1c). In addition, multiple, small, incipient tumors (under 0.5 cm in the largest dimension), which are also composed of clear cell RCC, are often scattered in the renal parenchyma (Fig. 6.1 d).
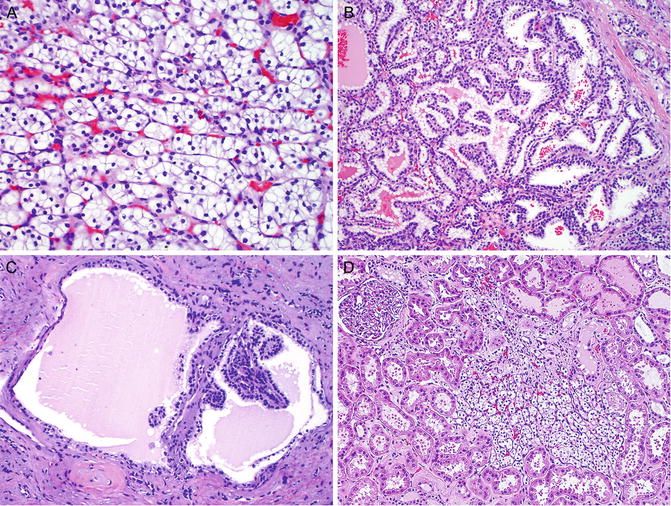
Fig. 6.1
Renal cell carcinoma in von Hippel-Lindau syndrome . The tumor is typically the clear cell type, which is characterized by small nests of tumor cells with clear cytoplasm separated by a delicate vascular network (a). Tumor cells may form papillae and tubules (b). Renal cysts are lined by a single layer of clear tumor cells, forming small tufts and papillae (c). Small tumors (less than 0.5 cm) of clear cell RCCs are common (d)
The immunohistochemical profile of VHL-associated RCC is similar to that of sporadic clear cell RCC. They usually show strong positive immunoreactivity for carbonic anhydrase IX (CAIX) and CD10 and are negative or focally positive for cytokeratin 7 (CK7). The reactivity for alpha-methylacyl-CoA racemase (AMACR ) is variable. Deletion of chromosome 3p is also common in VHL-associated RCC (82 %), at a level similar to that observed in sporadic clear cell RCC (80 %) [13].
VHL-associated RCC may have focal areas of tubular, papillary, and cystic structures lined by tumor cells with clear cytoplasm, which mimics clear cell papillary RCC (CCPRCC). CCPRCC is a new entity that has not been included in the current WHO classification [14]. However, unlike VHL-associated RCC, CCPRCC i s characterized by positive immunoreactivity for CK7 and negative reactivity for AMACR and CD10. Furthermore, while VHL-associated RCC frequently exhibits 3p deletion, CCPRCC lacks 3p deletion but may exhibit chromosome 7 or 17 abnormalities in a subset of cases [13].
Treatment
Historically many patients with VHL underwent radical nephrectomy, but renal parenchymal-sparing treatments are generally preferred in this and most other FRCC syndromes. Given the proclivity of most patients with VHL to form multiple and bilateral clear cell renal cell carcinomas over their lifetime, nephron-sparing should be entertained whenever oncologically feasible. Partial nephrectomy is now the standard extirpative approach to all but the largest renal tumors, and percutaneous ablation (with radiofrequency or cryotherapy) can also be offered to patients in lieu of radical nephrectomy unless the solid tumors have grown too large for such modalities [15]. A 3 cm size threshold prior to intervention for solid renal masses has been studied in order to limit the number of interventions in patients with some forms of FRCC such as VHL [16]. Data to date suggest that the metastatic potential of lesions less than 3 cm in diameter in VHL is close to zero, so solid renal masses in VHL, invariably clear cell RCC, are routinely observed until they grow. At that point, partial nephrectomy is performed if feasible, as well as enucleation of all smaller tumors and cysts that can be handled safely. If percutaneous ablation is chosen, only the 3 cm+ tumor is ablated and the smaller lesions are followed .
Hereditary Papillary Renal Cell Carcinoma (HPRCC)
Introduction
HPRCC is an inherited renal tumor syndrome characterized by an autosomal dominant trait with a high penetrance [17]. Affected individuals are at a high risk of developing a large number of tumors in both kidneys. This syndrome chiefly affects the kidney, and no extrarenal involvement has been reported to be associated with HPRCC. The tumors are invariably composed of papillary type 1 RCC. The onset of HPRCC is usually late in most patients, in the 6th to 8th decades of life, but a few cases of early onset have been reported [18].
HPRCC is commonly associated with germline mutations in the proto-oncogene MET , which is located at chromosome 7q31 [19]. MET encodes a transmembranous protein kinase, hepatocyte growth factor receptor [20]. Normally, hepatocyte growth factor receptor binds and activates MET tyrosine kinase, which in turn phosphorylates a number of signal transduction proteins. The MET tyrosine kinase plays an important role in cell proliferation and differentiation. Missense mutations in the MET tyrosine kinase domain lead to ligand-independent constitutive activation of the MET protein [21]. The aberrant activation of the MET protein leads to phosphorylation of signal transducers and adaptors, resulting in cancer development. MET somatic mutations have also been reported in some patients with sporadic papillary type 1 RCC [22].
Clinical Presentation
Solid papillary renal tumors are the only clinical manifestation of this form of FRCC, and therefore these tumors are picked up incidentally, by screening of families known to be affected or on workup for gross or microscopic hematuria. These tumors are invariably solid (though some cysts have been noted in affected individuals) and enhance only minimally compared to other solid renal tumors: +10–30 Hounsfield units on contrast CT or +15 % on contrast MRI [23]. No mature routine screening recommendations for mutation carriers exist, though abdominal imaging by CT or MR is generally recommended every 2 years .
Pathology
Typically, there are numerous tumors in bilateral kidneys, which may number in the hundreds to thousands on gross examination. Histologically, the renal tumors are composed of papillary type 1 RCC, which is characterized by papillae lined with a single layer of small tumor cells with scant amphophilic cytoplasm and low nuclear grade (Fig. 6.2a). Papillae may coalesce to impart solid growth patterns with glomeruloid structures (Fig. 6.2b). There is a variable amount of foamy histiocytes and psammomatous calcification in the papillary cores (Fig. 6.2c). Necrosis and hemosiderin deposits may be found. Clear cell changes may be present, especially in areas adjacent to necrosis. A large number of small papillary renal neoplasia that are identical to sporadic renal papillary adenomas are frequently seen in the renal parenchyma (Fig. 6.2d).
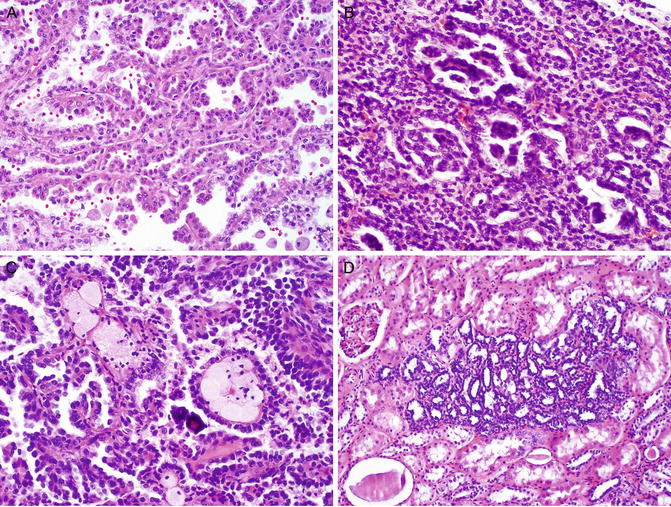
Fig. 6.2
Hereditary papillary renal cell carcinoma. The tumor is invariably a papillary type 1 RCC, which is characterized by papillae lined with a single layer of tumor cells with low nuclear grade (a). Papillae may coalesce a solid growth pattern with glomeruloid structures (b). Foamy histiocytes and psammomatous calcification are observed in the papillary cores (c). A large number of renal papillary adenomas are frequently seen in the grossly normal renal parenchyma (d)
It is difficult, if not impossible, to distinguish HPRCCs from sporadic papillary type 1 RCCs based on pathologic analysis, as they share similar histologic and immunohistochemical features [24]. A diagnosis of HPRCC may be entertained when a patient presents with a large number of papillary type 1 RCC and a family history of renal tumor. The diagnosis of HPRCC can be further supported by identifying the germline MET mutations on DNA sequencing .
Treatment
Type I papillary RCCs typically grow slowly and have low metastatic potential. They are therefore generally managed observationally until the 3 cm threshold for intervention is reached, as for many other forms of FRCC. The options of radical nephrectomy, partial nephrectomy, and percutaneous ablation are similarly offered for these tumors.
Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)
Introduction
HLRCC is an autosomal dominant cancer syndrome with a high penetrance [25]. These patients are at risk of developing papillary type 2 RCC, cutaneous and uterine leiomyomas. In affected patients, the penetrance of RCC is approximately 20–30 %, whereas the penetrance of cutaneous and uterine leiomyomas is much higher: 76 % of individuals and 100 % of women at a mean age of 25 years or 30 years [25–27]. Occasionally patients may develop leiomyosarcoma in the uterus. The median age of patients at onset of RCC is 36–44 years, which is much earlier than when sporadic kidney cancer manifests. The onset of leiomyomas occurs at a relatively young age: at 10–47 years for cutaneous leiomyomas and 18–52 years for uterine leiomyomas.
The gene responsible for HLRCC is fumarate hydratase (FH), a tumor suppressor which is located on chromosome 1 (1q42.3-q43) [25–27]. Germline mutations in FH gene have been found in 85 % of the HLRCC patients. The majority of germline mutations are missense mutations, although truncation and whole-gene deletion may occur. A “second hit” or somatic inactivation of the remaining FH allele is usually required to cause functional loss of FH protein. Because the FH protein regulates the conversion of fumarate to malate in the citric acid (Krebs) cycle, the inactivation of this protein increases the level of fumarate, which competitively inhibits the HIF prolyl hydroxylase [28]. Subsequently, the HIF level increases, influencing the expression of the downstream genes. There is no evidence to support a relationship between the FH gene mutation and sporadic RCC.
Clinical Presentation
This is a disorder with cutaneous, uterine, and renal manifestations, only the latter being cancerous. Patients generally develop cutaneous leiomyomata (trunk and extremity) in their 20s, and these can be painful. Females almost always develop multiple, large uterine leiomyomata during their young adulthood, which typically become quite symptomatic though rarely cancerous. In addition, some 10–16 % of affected patients also develop papillary RCC type 2 renal tumor [29]. These cancers are very aggressive, highly malignant, and quite different in behavior from most other papillary RCC and even from many clear cell RCC. Unlike other forms of FRCC, these tumors grow so rapidly that a patient will often present with just one large (and symptomatic) tumor despite their hereditary predisposition to multiple and bilateral tumors.
Pathology
Renal Cancer
On gross examination, HLRCC tumors are typically solitary and unilateral, unlike tumors of other hereditary RCC syndromes, which are characterized by bilateral distribution of multiple renal tumors. HLRCC tumors are usually large, tan, and firm with necrosis. Microscopically, HLRCC usually exhibits a large number of papillae covered by large tumor cells with abundant eosinophilic cytoplasm (Fig. 6.3 a ), as is seen in papillary type 2 RCC. The most characteristic feature of HLRCC is the presence of a large nucleus with a very prominent inclusion-like nucleolus that is surrounded by a clear halo, resembling cytomegalovirus nuclear inclusions (Fig. 6.3b). The tumor may also have solid, tubular, or tubulopapillary growth patterns (Fig. 6.3c). The tumor is often associated with desmoplastic and cystic changes, mimicking collecting duct carcinoma (CDC), but CDC lacks the characteristic nucleolar features of HLRCC. Furthermore, CDC is positive for CK7 and ULEX on immunostaining, while HLRCC is often negative or only focally positive for CK7 and negative for ULEX-1. Occasionally, the tumor may develop sarcomatoid dedifferentiation (Fig. 6.3d). The tumors often present at an advanced stage with invasion into renal vein, renal sinus, and perinephric soft tissue [30]. Recent studies have shown that the presence of S-(2-succino)-cysteine (2SC) positivity by immunohistochemical analysis predicts genetic alterations of the FH gene in patients and that this immunoreactivity is generally absent in non-HLRCC-related tumors [31, 32].
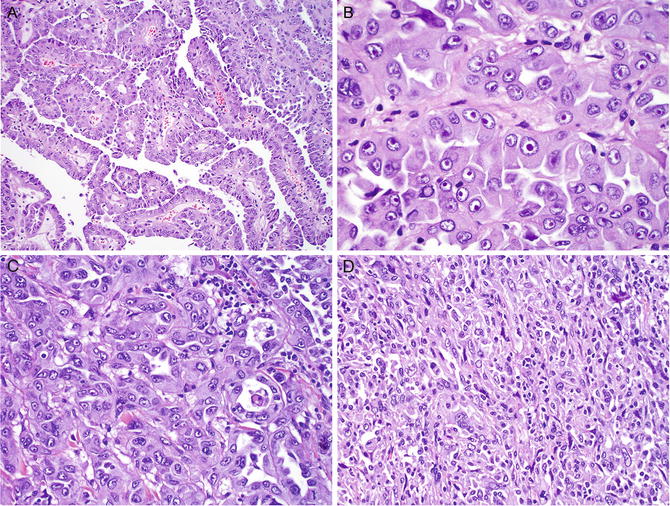
Fig. 6.3
Hereditary leiomyomatosis and renal cell carcinoma. The tumor typically appears like papillary type 2 RCC, which is composed of papillae covered by large tumor cells with a high nuclear grade and abundant eosinophilic cytoplasm (a). The most characteristic feature is the presence of a large nucleus with a prominent inclusion that is surrounded by a clear halo (b) . The tumor may have solid, tubular, or tubulopapillary growth patterns (c). The RCC may develop focal sarcomatoid dedifferentiation (d)
Leiomyomas of the Skin and Uterus
Cutaneous leiomyomas often appear as multiple, firm nodules ranging from 0.2 to 2.0 cm in the largest dimension. Microscopically, cutaneous leiomyomas are composed of anastomosing bundles of smooth muscle cells with inconspicuous nuclei. Uterine leiomyomas often present as numerous, well-circumscribed, and firm masses. Histologically, most leiomyomas are composed of whorled, interlacing fascicles of uniform, fusiform smooth muscle cells. A small number of patients develop uterine leiomyosarcomas [33], which are usually large and fleshy with poorly defined margins. The leiomyosarcomas are usually hypercellular with conspicuous nuclear atypia and increased mitotic activity.
Treatment
Prompt surgical management is recommended for HLRCC patients with a renal mass of any size. Unlike other forms of FRCC, where it is generally prudent to wait until a renal tumor grows to 3 cm prior to intervening, HLRCC renal tumors are treated on diagnosis. While there are not enough data to recommend nephron-sparing vs. radical nephrectomy for these tumors, prompt and aggressive treatment is warranted for these patients. Unfortunately, given that this syndrome has only recently been characterized, more than half of patients already had locally advanced or metastatic disease at presentation in the initial series [34].
Birt-Hogg-Dubé (BHD) Syndrome
Introduction
BHD syndrome is a rare autosomal dominant cancer syndrome with incomplete penetrance [35]. Renal tumors occur in 14–34 % of affected individuals with an onset commonly in the 6th decade of life (ranging 31–73 years). The majority of the renal tumors that develop are hybrid oncocytic tumors (50 %), chromophobe RCCs (33 %), or oncocytomas (5 %), but clear cell and papillary RCCs have occasionally been seen as well [36]. More than half of patients with BHD syndrome also have multifocal oncocytosis in the surrounding renal parenchyma [36].
BHD syndrome is caused by germline mutations in the folliculin gene (FLCN) [37], which is located at chromosome 17p11.2 [38]. These mutations can be insertions, deletions, or nonsense mutations. The FLCN gene, which functions as a tumor suppressor gene, requires mutations in both alleles to be inactivated. Usually, one allele has a germline mutation and the other allele has a somatic mutation. FLCN protein forms a complex with interacting proteins, which regulates the mammalian target of rapamycin (mTOR) signaling pathway via AMP-activated protein kinase (AMPK) [39]. Mutations in the FLCN gene may also be involved in the development of a number of sporadic RCCs [40].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


