Urinary Tract Tuberculosis
Mark S. Pasternack
Robert H. Rubin
Over a century ago, Osler noted that tuberculosis is the net result of two pathologic processes: “In all tubercles two processes go on: the one—caseation— destructive and dangerous; and the other—sclerosis— conservative and healing. The ultimate result in a given case depends upon the capabilities of the body to restrict and limit the growth of the bacilli.” 1
Perhaps in no other form of tuberculosis are these processes so important in determining the impact of the tubercle bacillus on an organ system as in genitourinary tuberculosis. Progressive destruction and caseating necrosis of the kidney ultimately leading to “autonephrectomy” have long been recognized as possible catastrophic complications of renal tuberculosis. However, progressive ureteral and calyceal scarring seen during healing can result in severe obstructive uropathy and comparable loss of renal function. Therefore, medical and surgical management of genitourinary tuberculosis must focus on both aspects of the tuberculous process, with emphasis on the early diagnosis and prevention of both tissue damage and excessive scarring. Achieving these goals can be challenging because genitourinary tract tuberculosis can be a particularly occult process. This chapter outlines a practical approach to this problem, based on established epidemiologic, pathogenetic, and clinical principles.
ETIOLOGY
Robert Koch first identified the tubercle bacillus in 1882. His classic report defined staining procedures for the direct observation of bacilli in clinical specimens (including the use of aniline dyes for “acid-fastness”), culture techniques on solid medium for the in vitro passage of bacilli isolated from clinical or experimental lesions, and subsequent inoculation of guinea pigs with cultured material to confirm its etiologic role in tuberculosis. 2 Demonstrating an etiologic role for the tubercle bacillus in tuberculosis became the basis of “Koch’s postulates,” the standard criteria for etiologic research in infectious disease.
Mycobacterium tuberculosis, the human tubercle bacillus, is one of approximately 90 species of higher bacteria with unusual shared structural and tinctorial properties. 3,4 All mycobacteria, members of the genus Mycobacterium, have the ability to take up aniline dyes, such as those contained in carbolfuchsin, and to resist decolorization by washing in alcohol acidified with inorganic acid (e.g., 95% ethanol, 3% HCl). This unique property correlates with the extremely high lipid content of mycobacterial cell walls. Although all mycobacteria are obligate aerobes, they are found in nature in disparate settings: some species are soil and water saprophytes, whereas others are true pathogens of amphibians, reptiles, birds, and various mammals. M. bovis, the bovine tubercle bacillus, has virtually disappeared as a human pathogen in modern societies through tuberculin testing of cattle and pasteurization of dairy products. A variety of mycobacterial species can be pathogenic in humans (e.g., M. avium-intracellulare), whereas others have been characterized as human saprophytes ( M. gastri, M. smegmatis ). M. tuberculosis is distinguished from the many other “atypical” mycobacteria by its metabolic properties, rate of growth, pigment production, and virulence in experimental infection in guinea pigs, as well as by genomic features that facilitate direct speciation. M. tuberculosis characteristically appears as a small, slender, slightly curved rod 2 to 4 µm in length with a diameter of 0.3 to 0.6 µ m. Bacilli can appear singly or in small clusters on clinical specimens. Unlike infected pulmonary secretions where the density of organisms commonly is high, the low density of bacilli in urine samples, as well as their possible confusion with saprophytic mycobacteria, makes urine acid-fast stains impractical for rapid diagnosis. Although M. tuberculosis can grow on simple synthetic media, typically in intertwining aggregates known as serpentine cords, its slow growth rate (15 to 20 hours doubling time) necessitates culture periods of up to 6 to 8 weeks for the appearance of visible colonies. Optimal growth requires high partial pressures of oxygen, as in air, although bacilli can remain viable but metabolically dormant under greatly reduced PO2 . This is particularly relevant for the progression of renal tuberculosis (see later).
The mycobacterial cell wall accounts not only for acid-fast staining, but for some of the important host-parasite
interactions as well. In addition to a peptidoglycan cell wall layer common to conventional bacteria, a second glycan layer encases the organism. 5,6 This arabinogalactan layer is covalently linked to the peptidoglycan layer and also contains esters of mycolic acids, which are very large fatty acids that are unique to mycobacteria. A number of complex glycolipids reside in the outermost layer—“cord factor” (trehalose dimycolate), phosphatides, and sulfatides—but are not covalently linked to the glycan layers. 6 Cord factor is responsible for growth in serpentine cords in vitro and is a virulence factor in vivo. 7 These cell wall moieties (lipoarabinomannan, trehalose dimycolate and its sulfated derivatives) have multiple effects on mycobacterial virulence: they inhibit phagosome maturation and fusion with lysosomes, 6,8 reduce cell surface expression of key host antigen presentation proteins and costimulatory molecules, thus diminishing the presentation of mycobacterial antigens by infected macrophages, 9 and even modulate macrophage survival and apoptosis. 10
interactions as well. In addition to a peptidoglycan cell wall layer common to conventional bacteria, a second glycan layer encases the organism. 5,6 This arabinogalactan layer is covalently linked to the peptidoglycan layer and also contains esters of mycolic acids, which are very large fatty acids that are unique to mycobacteria. A number of complex glycolipids reside in the outermost layer—“cord factor” (trehalose dimycolate), phosphatides, and sulfatides—but are not covalently linked to the glycan layers. 6 Cord factor is responsible for growth in serpentine cords in vitro and is a virulence factor in vivo. 7 These cell wall moieties (lipoarabinomannan, trehalose dimycolate and its sulfated derivatives) have multiple effects on mycobacterial virulence: they inhibit phagosome maturation and fusion with lysosomes, 6,8 reduce cell surface expression of key host antigen presentation proteins and costimulatory molecules, thus diminishing the presentation of mycobacterial antigens by infected macrophages, 9 and even modulate macrophage survival and apoptosis. 10
EPIDEMIOLOGY
Tuberculosis and HIV infection are each responsible for approximately 1.8 million deaths worldwide each year (which includes ˜400,000 deaths due to dual infections, especially in resource-limited settings). 11 The World Health Organization (WHO) estimates that approximately one third of the world’s population is latently infected with M. tuberculosis with approximately 9 million new cases occurring each year. Although more than 90% of cases occur in the developing world with significant overlap among symptomatic HIV infected individuals, 12 10 to 15 million individuals in the United States are infected with M. tuberculosis, mostly with latent tuberculosis infection. 13,14
The long-term secular decline in tuberculosis incidence that followed the development of successful antituberculous chemotherapy was disrupted in the 1980s by a decline in the support of tuberculosis control programs, as well as the interrelated challenges of the HIV epidemic, troubling social trends producing growing populations of homeless and incarcerated individuals, and the rising incidence of drug-resistant tuberculosis infections. 15,16,17 These processes often acted in synergy to produce epidemics of tuberculosis, frequently involving multiply drug-resistant strains, among vulnerable populations in hospitals, correctional facilities, residential care facilities, and homeless shelters. 18,19 The number of reported U.S. tuberculosis cases in 1992 (˜26,000) roughly equaled those of 1982, erasing a decade of progress in tuberculosis control. An intensification of tuberculosis control measures, including greater emphasis on intensive initial empiric therapy, sensitivity testing of clinical isolates, and reliance on directly observed therapy, has helped to regain control of the tuberculosis epidemic, and by 2010 only 11,181 new cases were reported. 20
Tuberculosis in the United States is primarily an urban disease with 75% of the new cases occurring in the 99 metropolitan areas that have populations of more than 500,000. Increasingly, in the United States tuberculosis is found among immigrants and minority groups. Approximately 80% of reported cases in 2010 occurred in Asian, Hispanic/Latino, and African American residents in roughly comparable numbers, whereas whites accounted for only 16% of cases. The nationwide incidence of tuberculosis was 3.6/100,000 overall, with marked variation by ethnicity and immigrant status: the incidence was highest among Asians and Pacific Islanders (22.5/100,000) and was lowest among non-Hispanic whites (0.9/100,000). In 2010 only 40% of cases occurred in native-born individuals, and in many states over 70% of cases occurred among foreign-born individuals. Dual infection with HIV was reported in approximately 10% of cases. 20 In Europe, similar patterns of increased tuberculosis have been seen among immigrants from high tuberculosis incidence countries. 21,22,23 In addition, relatively high rates of extrapulmonary tuberculosis and drug-resistant tuberculosis have been observed among these immigrant populations. 24,25
Although the proportion of extrapulmonary disease has nearly tripled from 7.6% to 21% of reported cases of tuberculosis in the United States over the past 40 years, the relative incidence of genitourinary infections among all forms of extrapulmonary tuberculosis has gradually declined. 26 Regional lymph node infections remain the most commonly encountered form of extrapulmonary tuberculosis— genitourinary disease, once common, 27 has declined to 6.5% of extrapulmonary tuberculosis cases in the United States over the past 20 years or so, a frequency roughly comparable to that of tuberculous meningitis. 26 Similar low rates of genitourinary tuberculosis have been reported recently from both low incidence (France) 28 and high incidence countries (Nepal). 29 As outlined later and in Figure 27.1, extrapulmonary tuberculosis is the result of hematogenous spread from a pulmonary site of primary infection. Thus, genitourinary tuberculosis is observed in two clinical settings: commonly, as a late manifestation of earlier clinical or subclinical pulmonary infection and, rarely, as part of the multiorgan infection seen with disseminated (miliary) tuberculosis.
Statistics from the prechemotherapy era indicated that approximately 3% of unselected autopsy patients and 26% of those dying of tuberculosis had evidence of genitourinary tract tuberculosis at autopsy. 30 This high rate of genitourinary disease has declined with effective treatment of pulmonary tuberculosis. Currently, it is estimated that significant genitourinary disease will develop in approximately 4% to 8% of non-HIV-infected individuals with pulmonary tuberculosis if adequate therapy is not instituted. 31
Traditionally, genitourinary tuberculosis has been a disease of young to middle-aged adults with a slight male predominance.27,31,32,33,34 Although genitourinary tuberculosis has been reported in children, 35,36,37 it is quite uncommon, and seen today in the rare young child with concomitant miliary disease 37 or in school-age children with somewhat more indolent clinical features similar to those seen in adults. 36 Approximately one quarter of the patients with genitourinary tuberculosis have a history of diagnosed tuberculosis
(usually of the lung). In an additional 25% to 50% of patients, changes compatible with old pulmonary tuberculosis can be found on chest X-ray films made at the time of diagnosis of genitourinary tract disease. 27,31,32,33
(usually of the lung). In an additional 25% to 50% of patients, changes compatible with old pulmonary tuberculosis can be found on chest X-ray films made at the time of diagnosis of genitourinary tract disease. 27,31,32,33
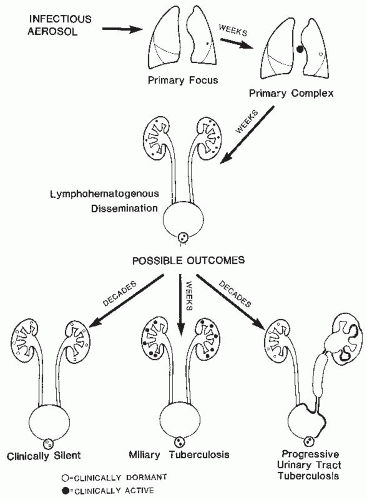 FIGURE 27.1 Schematic representation of the pathogenesis of urinary tract tuberculosis. |
Thus, in the non-HIV-infected individual a considerable interval exists between the onset of pulmonary infection and the diagnosis of active genitourinary tuberculosis. Considering patients with early clinical manifestations of primary tuberculous infection (i.e., erythema nodosum, pleurisy, or hilar adenopathy), the time lapse is most commonly 16 to 25 years; intervals exceeding 40 years have been well documented. If one looks at patients with reactivation pulmonary tuberculosis, the time lapse is usually about 4 to 8 years, but still may be as long as several decades.31,32,38
There are several epidemiologic implications of these observations. Decreasing the incidence of genitourinary tuberculosis requires the identification and treatment of persons with pulmonary infection prior to the development of extrapulmonary disease. Second, long after the incidence of pulmonary tuberculosis falls, the incidence of genitourinary disease will remain relatively stable, because a reservoir of patients with silent genitourinary tract infection will persist for decades after the incidence of new pulmonary infection falls. 24,25,26 Thus, the rise in tuberculosis cases that occurred in the late 1980s and early 1990s virtually guarantees an increase in extrapulmonary infections such as genitourinary disease in the next few decades, unless extensive case finding and effective treatment of these individuals are achieved. This task will be rendered particularly difficult because the burden of disease has fallen heavily on those with poorer access to skilled medical care—foreign born, the homeless, the incarcerated, migrant farm workers, and inner city ethnic minority groups. 16,17,20,39 Finally, it is clear that the age of the population with genitourinary tract tuberculosis, and other forms of extrapulmonary tuberculosis, reflects the average age at presentation of active pulmonary tuberculosis— significantly younger among immigrants, urban ethnic minority populations, and the disadvantaged, and over the age of 50 among other groups. 25
In assessing epidemiologic factors that might predispose to tuberculosis, it is important to emphasize that fewer than 10% of persons with latent tuberculosis infection (reflecting a positive tuberculin skin test or in vitro interferon-γ release assay) ever become ill from this infection. 40 Of this group,
approximately 3% to 5% have manifestations of genitourinary tract disease. 27 In addition to exposure to tuberculosis in high incidence countries, public health factors including crowding, homelessness, poverty, drug addiction, and incarceration, all amplified by the AIDS epidemic, 12 play a significant role in the spread of tuberculosis.
approximately 3% to 5% have manifestations of genitourinary tract disease. 27 In addition to exposure to tuberculosis in high incidence countries, public health factors including crowding, homelessness, poverty, drug addiction, and incarceration, all amplified by the AIDS epidemic, 12 play a significant role in the spread of tuberculosis.
A minimum estimate of clinical tuberculosis in HIV-infected patients is approximately 15% worldwide 11 and 8.6% in the United States, 20 with a somewhat higher incidence (13.8%) in adults 25 to 44 years of age. 41 The annual rate of tuberculosis among tuberculin skin test positive HIV-infected individuals in the United States has been reported to be 35 to 162 cases per 1,000 person years, although in Africa the risk of clinical tuberculosis may be as high as 5% to 10% annually. 42 These variations reflect differences in the prevalence of tuberculosis and immunodeficiency in different population groups. Thus, the rate of tuberculous disease among HIV-infected, tuberculin positive individuals in the United States is roughly 10 times higher than that among comparable HIV-infected, tuberculin negative individuals, 43 and is approximately 80 to 370 times that of the general American population. 44 The continued refinement and availability of antiretroviral therapy combined with antituberculous therapy has altered the natural history of tuberculosis in HIV-infected individuals and, when combined with vigorous public health efforts to identify and monitor treatment, the prognosis of both HIV and tuberculosis in dual-infected patients has improved dramatically. 12,45,46,47 Dual therapy is often challenging due to drug interactions: for example, rifabutin is required instead of rifampin if a protease inhibitor is used. Fluconazole metabolism is increased in the presence of rifampin48 and other agents, thus complicating care in these patients. Despite the need for care in constructing the therapeutic regimen, early diagnosis and appropriate therapy of both infections are critical to the patient’s survival. 41,44,46,47
Two epidemiologic patterns of mycobacterial infection are observed in HIV-infected individuals. Individuals from population groups with a low rate of endemic tuberculosis, such as gay men and those with posttransfusion HIV disease, primarily have difficulties with disseminated M. avium-intracellulare infection. In contrast, HIV-infected individuals who either belong to or interact with populations bearing a high rate of endemic tuberculosis (those in developing countries, immigrants from these countries, the homeless, intravenous drug users, prisoners, and, in the United States and other developed countries, the inner city poor) are primarily afflicted with disseminated M. tuberculosis . 44,45 Careful molecular epidemiologic studies have demonstrated that in HIV-infected individuals, outbreaks of tuberculosis resulted in either progressive primary disease or reinfection of individuals whose immunity had been attenuated by the effects of progressive HIV disease. 49,50
In HIV-infected individuals tuberculosis is often the AIDS-defining illness, not infrequently occurring early in the course of HIV infection. In contrast to other AIDS-related opportunistic infections, the CD4+ count is not a reliable indicator of tuberculosis risk among HIV-infected persons. 44 Extrapulmonary disease, often in conjunction with pulmonary disease, is common, 44 and the time course for the development of disseminated disease may be greatly abbreviated in these individuals. In this setting, genitourinary disease is less commonly seen as an isolated phenomenon, but rather as part of disseminated infection. The incidence of HIV infection among patients with extrapulmonary tuberculosis was significantly elevated in the initial phases of the HIV epidemic, 51 but at present the rate of extrapulmonary tuberculosis in dually infected individuals is comparable to that seen in HIV-uninfected patients. 26 Serial New York City data confirmed aggressive dual therapy led to a reduced rate of extrapulmonary tuberculosis in dually infected individuals. 45
The coexistence of tuberculosis with HIV infection has already had major public health consequences on the control of tuberculosis. Because HIV infection both increases the burden of infectious tubercle bacilli and obscures the symptoms (owing to the impaired inflammatory response of these immunosuppressed individuals), these people are highly efficient transmitters of tuberculosis. The expected consequence of this is the potential for a marked increase in the occurrence of secondary cases, and even epidemics, in contacts of these individuals, particularly in medical settings, crowded living conditions, prisons, and shelters for the homeless. Those populations most at risk for both HIV infection and tuberculosis, and the coexistence of these two infections, are the same populations with the highest incidence of drug-resistant tuberculosis. The result of this concordance of events is that the tuberculosis that can be amplified by the HIV epidemic includes a high potential for drug-resistant disease, particularly in the former states of the Soviet Union and more generally in resource-limited settings, and can complicate both the management of individual patients and the public health strategies that must be taken to protect the community. 11
One additional epidemiologic consideration is the possibility that urine from patients with urinary tract tuberculosis could transmit tuberculosis to household members. Vasquez and Lattimer 52 reported a doubling of the incidence of tuberculin positivity among children of parents with active urinary tract tuberculosis without active pulmonary disease. Other observers have been unable to confirm this finding. Our policy has been not to isolate persons with isolated urinary tract tuberculosis but to consider their urine potentially infectious and to maintain contact precautions when handling it.
PATHOGENESIS OF TUBERCULOSIS
Systemic Aspects
The host-pathogen interaction in tuberculosis involving a slowly proliferating pathogen that resists host microbicidal mechanisms stands in stark contrast to conventional bacterial disease, where despite the pathogen’s rapid proliferation, the host’s resources (complement fixation, opsonization, phagocytosis, and ready lysis within phagocytic cells)
are formidable. The pathogenesis of tuberculosis reflects the balance of intrinsic mycobacterial virulence and the host immunologic response. The classic response in tuberculosis, the formation of granulomas, is ordinarily protective for the host by limiting the proliferation and spread of M. tuberculosis, but may be pathogenic as well, because it may lead directly to tissue injury in the form of caseation. 53,54,55 Thus, the clinical manifestations of tuberculosis represent not only the consequences of mycobacterial proliferation but also host reparative and destructive responses. In the absence of immunosuppression, the lifetime risk of symptomatic M. tuberculosis infection among latently infected individuals is only ˜ 10%. Similarly, the risk of developing extrapulmonary disease, such as genitourinary tuberculosis, is rather low. An increased understanding of the molecular mechanisms responsible for host defense against mycobacterial infections has led to a growing appreciation that human susceptibility to mycobacterial infection may be attributable in significant measure to host genetic factors, as well as to the intrinsic virulence of M. tuberculosis isolates. 56,57,58
are formidable. The pathogenesis of tuberculosis reflects the balance of intrinsic mycobacterial virulence and the host immunologic response. The classic response in tuberculosis, the formation of granulomas, is ordinarily protective for the host by limiting the proliferation and spread of M. tuberculosis, but may be pathogenic as well, because it may lead directly to tissue injury in the form of caseation. 53,54,55 Thus, the clinical manifestations of tuberculosis represent not only the consequences of mycobacterial proliferation but also host reparative and destructive responses. In the absence of immunosuppression, the lifetime risk of symptomatic M. tuberculosis infection among latently infected individuals is only ˜ 10%. Similarly, the risk of developing extrapulmonary disease, such as genitourinary tuberculosis, is rather low. An increased understanding of the molecular mechanisms responsible for host defense against mycobacterial infections has led to a growing appreciation that human susceptibility to mycobacterial infection may be attributable in significant measure to host genetic factors, as well as to the intrinsic virulence of M. tuberculosis isolates. 56,57,58
Tubercle bacilli are inhaled as small particle aerosols and gain direct access to the alveoli. 53 Presently, ingestion of M. tuberculosis with primary localization of disease in the intestinal tract or oropharynx is rare. The small aerosol inoculum multiplies slowly and is phagocytosed by polymorphonuclear leukocytes, pulmonary macrophages, and dendritic cells. Mycobacteria interact with respiratory epithelium,59 alveolar surfactant proteins, 60 and both the classic61 and alternate complement systems, 62 but interactions with macrophage Toll-like receptors (TLRs 2, 4, and 9) 63,64,65 play a critical role in initiating the host immune response. TLRs are pattern-recognition proteins, expressed on macrophages and dendritic cells, which serve as innate immune receptors. 66 Each TLR binds one or more of a variety of microbial products (endotoxin [lipopolysaccharide], bacterial DNA, flagellin, mycobacterial lipoarabinomannan, etc.) and transduce inflammatory signals culminating in the activation of NF-κ B and transcription of tumor necrosis factor α (TNF-α) and interferon-γ. 67 Following opsonization by C3, M. tuberculosis binds to phagocytic cell surface complement receptors and is phagocytosed. 62 Mycobacteria suppress the intracellular calcium flux that normally accompanies phagocytosis and inhibits macrophage activation and phagolysosome maturation. 68,69 M. tuberculosis can also directly adhere to, infect, and translocate across alveolar epithelial cells and endothelial cells, 70,71,72 facilitating access to lung interstitium and the pulmonary microcirculation, enhancing early dissemination to extrapulmonary foci.
The host response to mycobacterial infection has been called the IL-12-interferon-γ axis. 56,58 Macrophage activation via TLR signaling and other early events is associated with secretion of TNF-α and IL-12 and the related cytokines IL-18 and IL-23 65,73 as well as by activation of NO synthase 2, leading to the synthesis of reactive nitrogen intermediates.65 The cytokines program resting T lymphocytes toward an inflammatory Th1 response. Activated Th1 lymphocytes secrete as their dominant cytokines interferon-γ as well as TNF-α, and this in turn activates macrophages and enhances their mycobactericidal activity. TNF-α also triggers apoptosis of infected macrophages, which may inhibit mycobacterial replication (see later). The use of TNF-α antagonists (e.g., etanercept, infliximab, and adalimumab) as diseasemodifying agents in the treatment of rheumatoid arthritis and other inflammatory diseases confirms a central role for TNF-α in the host response against mycobacterial infection. These therapies have been associated with rapidly progressive tuberculosis, impaired granulomatous reactions in tissue biopsies, and a high rate of extrapulmonary disease. 74
In the initial stages of primary infection, resting macrophages have a limited ability to lyse mycobacteria, and the bacillary titer rises despite entrapment within macrophage and granulocyte phagosomes and lysophagosomes. Some bacilli can even escape from these organelles and replicate freely within the cytoplasmic compartment. 75 Macrophage-mediated killing of intracellular M. tuberculosis requires the L-arginine-dependent generation of reactive nitrogen intermediates, such as nitric oxide, and this capability is greatly enhanced following macrophage activation by interferon-γ and TNF. 65,76 Thus, infected macrophages program T cells toward a Th1 response which in turn augments macrophage- mediated mycobacterial killing. Foamy macrophages are characteristically seen in caseating granulomata and offer a protected locus of mycobacterial persistence; the disruption of these macrophages helps to recycle M. tuberculosis to the extracellular milieu. 77 T cell-infected macrophage interactions are more complex because several cytolytic T cell effector populations are generated which can lyse infected macrophages. 78,79 In addition to the expansion of conventional peptide- specific CD4+ and CD8+ αβ T-cell receptor-expressing populations, 79 T cells with a double negative phenotype (CD4-, CD8-) expressing γδ T-cell receptors recognize mycobacterial phospholipid antigens presented by MHC-like molecules (e.g., CD-1) and lyse infected macrophages via Fas-FasL interaction. 80 These phospholipid antigens are also recognized by CD8+ αβ T-cell receptor- expressing cytotoxic T lymphocytes, triggering perforin- mediated cytotoxicity, and by natural killer (NK) T cells, which possess NK markers as well as αβ T-cell receptors.81 In at least some instances, immune lysis of infected macrophages appears beneficial to the host. Perforinmediated lysis of infected macrophages reduces M. tuberculosis viability by 50% in vitro, 81 and may be important in vivo in reducing the number of infecting tubercle bacilli. The cytotoxic granules that contain perforin also contain granulysin, a lipid-binding protein that has potent mycobactericidal activity in the presence of perforin. 82 In contrast, Fas-mediated lysis of macrophages does not affect mycobacterial viability, but may be important in reducing antigen presentation and dampening the immune response. The immune lysis of heavily infected macrophages may facilitate the phagocytosis of released mycobacteria by additional activated macrophages. These newly recruited cells have a lower bacillary
burden and may be more effective at killing their intracellular bacilli, or serve as target cells for cytotoxic T lymphocytes.
burden and may be more effective at killing their intracellular bacilli, or serve as target cells for cytotoxic T lymphocytes.
In addition to their direct cytotoxic activities, the activated lymphocytes secrete a variety of cytokines, including interferon-γ, migration inhibitory factor (MIF), granulocyte-macrophage colony-stimulating factor, TNF-α, IL-12, and inhibitory cytokines such as interleukin-4 (IL-4) 83 and IL-10. 84,85 IL-4 production undermines the host Th1 response, and triggers tissue fibrosis, 86 a characteristic finding in chronic tuberculosis. In HIV-infected individuals coinfected with M. tuberculosis, immunosuppressive cytokines (e.g., IL-10) produced by macrophages/monocytes diminish the T-lymphocyte response in vitro, suggesting that Th2-like activity contributes to uncontrolled, systemic spread in these patients. 87 Macrophages are recruited to infiltrate the area of mycobacterial growth to form granulomas and mature into epithelioid cells by the macrophages’ elaboration of TNF 88 ; this process requires NK T cells. 89 In spite of macrophage activation, the killing of intracellular mycobacteria by human macrophages is often incomplete. 69
The outcome of early tuberculous disease covers a spectrum from granuloma formation with efficient containment and healing to slowly progressive disease at the site of the primary pulmonary infection, or to clinically significant systemic spread of disease. Mycobacterial dissemination is actually the rule rather than the exception (Fig. 27.1). Although most bacilli are contained within macrophages initially, their continued proliferation disrupts the macrophages and the bacilli return to the extracellular environment. Most are engulfed again, but some bind to respiratory epithelial cells and ultimately translocate to the microcirculation 71,72 or are carried in the lymphatic drainage and produce regional lymphadenitis. Alternatively, some viable mycobacteria may reach regional lymph nodes while entrapped within dendritic cells or migratory macrophages. Progressive infection within the lymph node contaminates efferent lymph, and when sequential lymph node barriers fail, thoracic duct lymph delivers mycobacteria to the venous blood, seeding the pulmonary bed as well as extrapulmonary sites, such as the skeletal system, lymph nodes, and, most frequently, the kidneys.
Thus, limited hematogenous dissemination due to low grade bacillemia can occur early in the process of granuloma formation when the number of mycobacteria is small, and most organisms are found intracellularly within the macrophages comprising the granuloma. Small granulomas rapidly form at the metastatic foci because mycobacterial immunity is evolving or is already established at the time of dissemination. Although the bacilli may remain viable, the granulomas may remain clinically silent for decades.
Granuloma formation may itself contribute to the pathogenesis of severe tuberculosis. 53 Granulomas are active lesions with continued ingress of immune T lymphocytes and monocytes. 54,90 Shortly after microscopic granulomas become well established, polymorphonuclear leukocytes and monocytes enter the lesion. 91 The resultant phagocytosis is accompanied by exocytosis of lysosomal contents with local tissue destruction. This leads to a characteristic local necrotic process, known as caseation. Macrophage disruption returns the mycobacteria to the extracellular environment, where their proliferation accelerates. 53 Communication of the caseating granuloma with the bronchial tree restores favorable metabolic conditions, and mycobacterial titers can increase by several logarithms. This highly infected material can spread endobronchially to produce additional foci of pulmonary tuberculosis or excavate into a pulmonary vessel, leading to intense bacillemia. Such severe hematogenous dissemination commonly is responsible for miliary tuberculosis rather than limited extrapulmonary disease. In miliary tuberculosis, the systemic features of illness overshadow the asymptomatic renal involvement.
Pathogenesis of Renal Tuberculosis
Local factors play a significant role in the evolution of clinically significant renal tuberculosis. The small silent renal granulomas resulting from silent hematogenous dissemination are typically found bilaterally in the renal cortex and arise from capillaries within and adjacent to glomeruli (Fig. 27.2




Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
