Urinary Obstruction & Stasis: Introduction
Because of their potential damage on renal function, urinary obstruction and stasis are important urologic disorders. Ureteral obstruction leads to hydronephrosis, kidney atrophy that may terminate in renal failure. Furthermore, obstruction often is complicated by infection, which causes additional damage to the organs involved.
Classification
Obstruction may be classified according to cause (congenital or acquired), duration (acute or chronic), degree (partial or complete), and level (upper or lower urinary tract).
Congenital anomalies, more common in the urinary tract than in any other organ system, are generally obstructive. In adult life, many types of acquired obstructions can occur.
The common sites of congenital narrowing are the external meatus in boys (meatal stenosis) or just inside the external urinary meatus in girls, the distal urethra (stenosis), posterior urethral valves, ectopic ureters, ureteroceles, and the ureterovesical and ureteropelvic junctions (Beganovic´ et al, 2007; Tan and Smith, 2004). Another congenital cause of urinary stasis is damage to sacral roots 2–4 as seen in spina bifida and myelomeningocele. Vesicoureteral reflux causes both vesical and renal stasis (see Chapter 13).
Acquired obstructions are numerous and may be primary in the urinary tract or secondary to retroperitoneal lesions that invade or compress the urinary passages. Among the common causes are (1) urethral stricture secondary to infection or injury; (2) benign prostatic hyperplasia or cancer of the prostate; (3) vesical tumor involving the bladder neck or one or both ureteral orifices; (4) local extension of cancer of the prostate or cervix into the base of the bladder, occluding the ureters; (5) compression of the ureters at the pelvic brim by metastatic nodes from cancer of the prostate or cervix; (6) ureteral stone; (7) retroperitoneal fibrosis or malignant tumor; and (8) pregnancy.
Neurogenic dysfunction affects principally the bladder. The upper tracts are damaged secondarily by ureterovesical obstruction or reflux and, often, by complicating infection. Severe constipation, especially in children, can cause bilateral hydroureteronephrosis from compression of the lower ureters.
Elongation and kinking of the ureter secondary to vesicoureteral reflux commonly lead to ureteropelvic obstruction and hydronephrosis. Unless a voiding cystourethrogram is obtained in children with this lesion, the primary cause may be missed and improper treatment given.
Obstruction and neuropathic vesical dysfunction have the same effects on the urinary tract. These changes can best be understood by considering the effects of (1) a severe meatal stricture on the lower tract (distal to the bladder neck), (2) a large obstructing prostate on the midtract (bladder), and (3) an impacted stone in the ureter on the upper tract (ureter and kidney).
Hydrostatic pressure proximal to the obstruction causes dilation of the urethra. The wall of the urethra may become thin, and a diverticulum may form. If the urine becomes infected, urinary extravasation may occur, and periurethral abscess can result. The prostatic ducts may become widely dilated.
In the earlier stages (compensatory phase), the muscle wall of the bladder becomes hypertrophied and thickened. With decompensation, it becomes less contractile and, therefore, weakened (Lieber et al, 2010).
To balance the increasing outlet resistance, the bladder musculature hypertrophies. Its thickness may double or triple. Complete emptying of the bladder is thus made possible.
Hypertrophied muscle may be seen endoscopically. With secondary infection, the effects of infection are often superimposed. There may be edema of the submucosa, which may be infiltrated with plasma cells, lymphocytes, and polymorphonuclear cells. At cystoscopy, surgery, or autopsy, the following evidence of this compensation may be visible (Figure 12–1):
Figure 12–1.
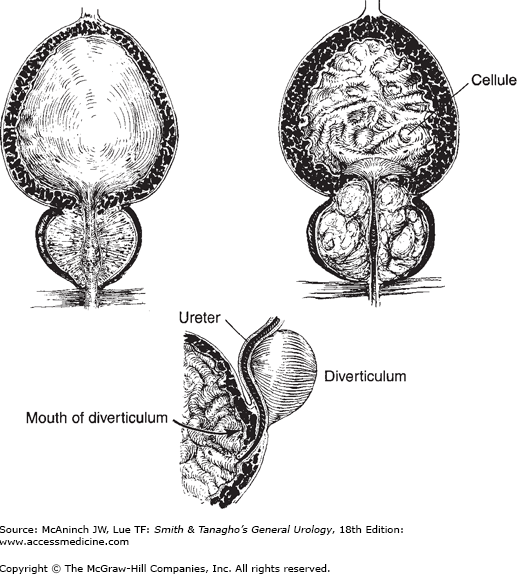
Changes in the bladder developing from obstruction. Upper left: Normal bladder and prostate. Upper right: Obstructing prostate causing trabeculation, cellule formation, and hypertrophy of the interureteric ridge. Bottom: Marked trabeculation (hypertrophy) of the vesical musculature; diverticulum displacing left ureter. (Redrawn and modified, with permission, from Tanagho EA, Pugh RCB: The anatomy and function of the ureterovesical junction. Br J Urol 1963;35:151.)
The wall of the distended bladder is normally quite smooth. With hypertrophy, muscle bundles with deposit of interstitial collage fibers become taut and give a coarsely interwoven appearance to the mucosal surface commonly described as trabeculation. The trigonal muscle and the interureteric ridge, which normally are only slightly raised above the surrounding tissues, respond to obstruction by hypertrophy of their smooth musculature. The ridge then becomes prominent. This trigonal hypertrophy causes increased resistance to urine flow in the intravesical ureteral segments. It is this mechanism that causes relative functional obstruction of the ureterovesical junctions, leading to back pressure on the kidney and hydroureteronephrosis. The obstruction increases in the presence of significant residual urine, which further stretches the ureterotrigonal complex (Tanagho and Meyers, 1965). (A urethral catheter relieves the obstruction somewhat by eliminating the trigonal stretch. Definitive prostatectomy leads to permanent release of stretch and gradual softening of trigonal hypertrophy with relief of the obstruction.)
Normal intravesical pressure is about 30 cm of water at the beginning of micturition. Pressures two to four times as great may be reached by the trabeculated (hypertrophied) bladder in its attempt to force urine past the obstruction. This pressure tends to push mucosa between the superficial muscle bundles, causing the formation of small pockets, or cellules (Figure 12–1).
If cellules force their way entirely through the musculature of the bladder wall, they become saccules, then actual diverticula, which may be embedded in perivesical fat or covered by peritoneum, depending on their location. Diverticula have no muscle wall and are therefore unable to expel their contents into the bladder efficiently even after the primary obstruction has been removed. When secondary infection occurs, it is difficult to eradicate; surgical removal of the diverticula may be required. If a diverticulum pushes through the bladder wall on the anterior surface of the ureter, the ureterovesical junction will become incompetent (see Chapter 13).
In the presence of acute infection, the mucosa may be reddened and edematous. This may lead to temporary vesicoureteral reflux in the presence of a “borderline” junction. The chronically inflamed membrane may be thinned and pale. In the absence of infection, the mucosa appears normal.
The compensatory power of the bladder musculature varies greatly. One patient with prostatic enlargement may have only mild symptoms of prostatism but a large obstructing gland that can be palpated rectally and observed cystoscopically; another may suffer acute retention and yet have a gland of normal size on rectal palpation and what appears to be only a mild obstruction cystoscopically.
In the face of progressive outlet obstruction, possibly aggravated by prostatic infection with edema or by congestion, decompensation of the detrusor may occur, resulting in the presence of large amount of residual urine after voiding. The amount may range up to 500 mL or more.
In the early stages of obstruction, intravesical pressure is normal while the bladder fills and is increased only during voiding. The pressure is not transmitted to the ureters and renal pelves because of the competence of the ureterovesical “valves.” (A true valve is not present; the ureterotrigonal unit, by virtue of its intrinsic structure, resists the retrograde flow of urine.) However, owing to trigonal hypertrophy (see Section “Trabeculation of the bladder wall”) and to the resultant increase in resistance to urine flow across the terminal ureter, there is progressive back pressure on the ureter and kidney, resulting in ureteral dilatation and hydronephrosis. Later, with the phase of decompensation accompanied by residual urine, there is an added stretch effect on the already hypertrophied trigone that increases appreciably the resistance to flow at the lower end of the ureter and induces further hydroureteronephrosis. With decompensation of the ureterotrigonal complex, the valve-like action may be lost, vesicoureteral reflux occurs, and the increased intravesical pressure is transmitted directly to the renal pelvis, aggravating the degree of hydroureteronephrosis (Riccabona, 2010; Routh et al, 2010).
Secondary to the back pressure resulting from reflux or from obstruction by the hypertrophied and stretched trigone or by a ureteral stone, the ureteral musculature thickens in its attempt to push the urine downward by increased peristaltic activity (stage of compensation). This causes elongation and some tortuosity of the ureter (Figure 12–2). At times, this change becomes marked, and bands of fibrous tissue develop. On contraction, the bands further angulate the ureter, causing secondary ureteral obstruction. Under these circumstances, removal of the obstruction below may not prevent the kidney from undergoing progressive obstruction due to the secondary ureteral obstruction.
Figure 12–2.
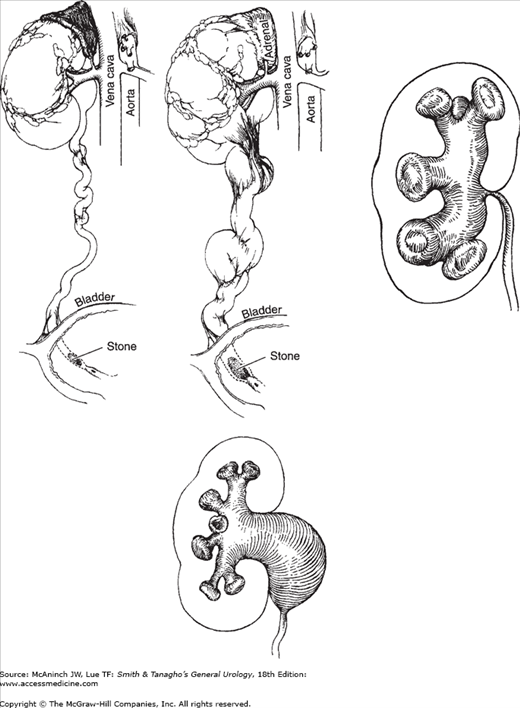
Mechanisms and results of obstruction. Upper left: Early stage. Elongation and dilatation of ureter due to mild obstruction. Upper center: Later stage. Further dilatation and elongation with kinking of the ureter; fibrous bands cause further kinking. Upper right: Intrarenal pelvis. Obstruction transmits all back pressure to parenchyma. Lower: Extrarenal pelvis, when obstructed, allows some of the increased pressure to be dissipated by the pelvis. (Reproduced, with permission, from Tanagho EA et al: Primary vesicoureteral reflux: Experimental studies of its etiology. J Urol 1965;93:165.)
Finally, because of increasing pressure, the ureteral wall becomes attenuated and therefore loses its contractile power (stage of decompensation). Dilatation may be so extreme that the ureter resembles a loop of bowel (Gimpel et al, 2010) (Figures 12–3 and 13–8, upper right).
Figure 12–3.
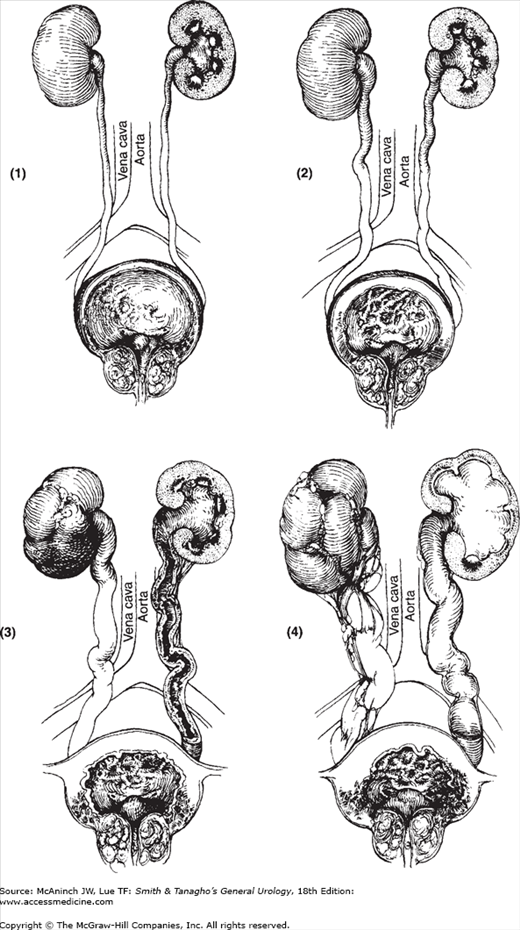
Pathogenesis of bilateral hydronephrosis. Progressive changes in bladder, ureters, and kidneys from obstruction of an enlarged prostate: thickening of bladder wall, dilatation and elongation of ureters, and hydronephrosis. (Reproduced, with permission, from Tanagho EA: Ureteroceles: Embryogenesis, pathogenesis and management. J Cont Educ Urol [Feb] 1979;18:13.)
The pressure within the renal pelvis is normally close to zero. When this pressure increases because of obstruction or reflux, the pelvis and calyces dilate. The degree of hydronephrosis that develops depends on the duration, degree, and site of the obstruction (Figure 12–4). The higher the obstruction, the greater the effect on the kidney. If the renal pelvis is entirely intrarenal and the obstruction is at the ureteropelvic junction, all the pressure will be exerted on the parenchyma (Klein et al, 2010). If the renal pelvis is extrarenal, only part of the pressure produced by a ureteropelvic stenosis is exerted on the parenchyma; this is because the extrarenal pelvis is embedded in fat and dilates more readily, thus “decompressing” the calyces (Figure 12–2).
In the earlier stages, the renal pelvic musculature undergoes compensatory hypertrophy in its effort to force urine past the obstruction. Later, however, the muscle becomes stretched and atonic (and decompensated).
The progression of hydronephrotic atrophy is as follows (Chevalier, 2010; Rodriguez, 2004):
The earliest changes in the development of hydronephrosis are seen in the calyces. The end of a normal calyx is concave because of the papilla that projects into it; with increase in intrapelvic pressure, the fornices become blunt and rounded. With persistence of increased intrapelvic pressure, the papilla becomes flattened, then convex (clubbed) as a result of compression enhanced by ischemic atrophy (Figure 12–5). The parenchyma between the calyces is affected to a lesser extent. The changes in the renal parenchyma are due to (a) compression atrophy from increase in intrapelvic pressure (more accentuated with intrarenal pelves) and (b) ischemic atrophy from hemodynamic changes, mainly manifested in arcuate vessels that run at the base of the pyramids parallel to the kidney outline and are more vulnerable to compression between the renal capsule and the centrally increasing intrapelvic pressure.
This spotty atrophy is caused by the nature of the blood supply of the kidney. The arterioles are “end arteries”; therefore, ischemia is most marked in the areas farthest from the interlobular arteries. As the back pressure increases, hydronephrosis progresses, with the cells nearest the main arteries exhibiting the greatest resistance.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree








