Fig. 25.1
Schematic representation of the blood supply of the adrenal glands
Radiology of Adrenal Gland
The major advantage of adrenal tumours is that it is often possible to reach the correct diagnosis pre-operatively, based on clinical assessment and non-invasive radiological and endocrinological investigations. This important feature allows safe and well-planned treatment strategies, with avoidance of unpleasant surprises. An expert radiologist is vital in sorting out renal from adrenal lesions and whether adrenal lesions are invading the adjacent kidney. The details of various radiological investigations are discussed in Chap. 4.
Computerised Tomography is the cornerstone of adrenal imaging and performed before and after intravenous injection of contrast medium [3]. On a normal CT, adrenals look homogeneous and symmetrical [3]. The combination of unenhanced CT and contrast washout values of adrenal masses can assist in characterisation and distinguishing adenomas from other adrenal tumours with 98 % sensitivity and 92 % specificity [3, 4].
Magnetic Resonance Imaging (MRI)
T1 and T2 weighted images with chemical shift imaging (CSI) is desirable with MRI of adrenals [3]. CSI relies on differentiating lesions by their lipid content, as malignant lesions do not have lipid in them. Multi-planar MRI helps in precise anatomical localisation and separation of adrenal masses from the surrounding structures, particularly the liver on the right side, spleen, stomach, pancreas and kidneys. Normal adrenal glands have T1 and T2 signal intensity equal or slightly lower than that of the normal liver [5].
Functional Imaging of Adrenal Cortical Masses
This involves imaging using isotopes based on physiological and pathological aspects of the adrenal gland. These investigations are particularly helpful when lesions are not adequately characterised by CT or MRI. At present 18F FDG PET seems an ideal method for characterising these tumours as benign or malignant [6]. Of those tumours that are malignant secondary tumours from a lung or gastro-intestinal primaries are more common than a primary adrenocortical tumour. Within the adrenal gland itself 18F FDG PET can be positive in both primary and secondary malignancy within the adrenal gland with the probable diagnosis depending on the pattern of activity and clinical history.
The most common functional benign adrenocortical tumour is the aldosterone producing Conn’s syndrome. This was traditionally imaged with 75Se methionine but this has been withdrawn from use. An alternative is 131I-iodocholesterol, but this is both very expensive and is associated with a very high radiation burden [7]. More recently a few centres have looked at 11C-metomidate PET-CT this allows rapid high quality imaging of Conn’s syndrome and can be used to find and characterise tumours as small as 4 mm [8].
Functional Imaging for Adrenal Medulla
Most pheochromocytomas express the noradrenaline transport system [9]. This transport system is responsible for the uptake of noradrenaline and adrenaline. The alkylguanidine meta-iodobenzylguanidine (MIBG), a noradrenaline analogue, is used in the diagnosis of pheochromocytoma as its cells take up MIBG. Imaging with 123I- or 131I-MIBG has been useful for characterising a suspicious adrenal gland in a patient with clinical suspicion of a pheochromocytoma and in staging patients with malignant pheochromocytomas where the advent of SPECT-CT allows even small lesions to be identified [10] (Fig. 25.2).
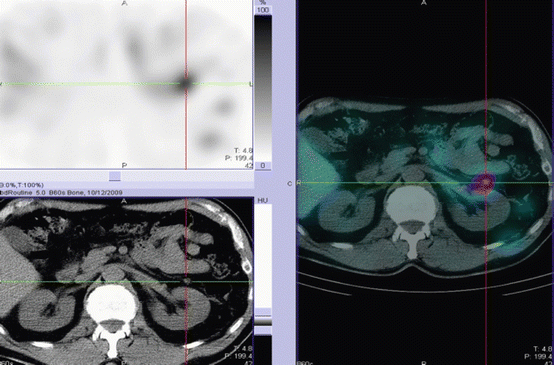
Fig. 25.2
A SPECT-CT 123I-mIBG image of the upper abdomen. Both the CT images and the SPECT image shows a small lesion in the post left adrenalectomy bed. However, only the combined SPECT-CT image confirms small volume residual tumour (Courtesy of Dr. John Buscombe, Royal Free Hospital, London)
There is also the possibility of treating patients with metastatic pheochromocytomas with high activity 131I-mIBG with good results obtained for both symptom relief and palliation of symptoms [11].
Adrenal Biopsy/Fine Needle Aspiration
With the advent of sophisticated radiological and biochemical investigations, there is a decreasing need for invasive tests such as needle biopsy or fine needle aspiration. If required the procedure is done with the CT guidance. Adrenal biopsy is not recommended unless it is part of staging a known malignancy [12]. Before biopsy, pheochromocytoma should be excluded with biochemical tests [13]. Because of its close proximity with diaphragm and pleura, potential complications include pneumo/hemothorax. Bleeding and infection are other complications.
A major problem in adrenal disease is that the glands frequently become nodular with age, presumably as part of a degenerative process, followed by a regenerative cycle. This tendency to nodularity is seen commonly in other endocrine glands, classically the thyroid. One task is to separate such natural nodularity from the diseases that require treatment. Another difficulty in adrenal pathology is distinguishing benign from malignant adrenal tumours by histological analysis. This lack of histological certainty means that careful clinical follow-up is often required.
Adrenal Incidentalomas
The term adrenal incidentaloma (AI) is usually defined as an adrenal mass unexpectedly detected through an imaging procedure performed for reasons a priori unrelated to adrenal dysfunction or suspected dysfunction [12]. Autopsy rates indicate a prevalence of incidental adrenal masses of 2–9 % increasing with age [14, 15]. CT scan studies have demonstrated detection rates of adrenal masses of 0.6–4.4 % [16]. Like renal tumours widespread application of abdominal imaging (ultrasound, CT and MRI) has resulted in increased frequency of clinically silent adrenal lesions. The majority are benign and non-functioning, but up to 20 % are functioning or malignant, and are therefore of great clinical significance.
Epidemiology
Prevalence of incidentalomas peaks between the ages of 50–60 years, with a slight female preponderance [17]. Incidentalomas are particularly common in patients with features of the metabolic syndrome (insulin resistance, dyslipidaemia, obesity and hypertension).
Causes of Incidentalomas
The differential diagnosis of adrenal incidentaloma is extensive but most of them are non-secreting cortical adenomas [12] (Table 25.1). Detection rates of incidentalomas on imaging and autopsy are shown in Table 25.2.
Table 25.1
Differential diagnosis of adrenal incidentaloma
Adrenal cortical tumours | Adenoma, carcinoma, nodular hyperplasia |
Adrenal medullary tumours | Pheochromocytoma, ganglioneuroma, neuroblastoma |
Other adrenal tumours | Myelolipoma, metastasis, hamartoma, teratoma, lipoma, hemangioma |
Infection | Bacterial: abscess, tuberculosis |
Fungal: histoplasmosis, coccidiomycosis, blastomycosis, | |
Viral: cytomegalovirus | |
Parasitic cysts | |
Granulomatous | Amyloidosis, sarcoidosis |
Cystic | Parasitic, endothelial, degenerative adenoma, haemorrhagic cyst |
Organs around adrenal | Renal, pancreatic, splenic, vascular |
Technical | Artefacts |
Table 25.2
Causes of incidentalomas detected by imaging and autopsy
Pathology | Rate of detection % (n = 267) |
|---|---|
Adrenal adenoma (non-functioning) | 86 |
Adrenal adenoma functioning (cortisol-secreting) | 8.6 |
Myelolipoma | 3 |
Pheochromocytoma | 2.6 |
Ganglioneurinoma | 1 |
Adrenocortical carcinoma | 0.3 |
Cysts | 0.022 |
Metastases | 0.01 |
Nodular hyperplasia | 0.01 |
Adrenal adenoma-functioning (aldosterone-secreting) | 0.003 |
Haemorrhage/haematoma | 0.003 |
Adrenal adenoma functioning (androgen-secreting) | 0 |
Other- lymphoma, congenital adrenal hyperplasia | 0 |
History and Physical Examination
A thorough clinical assessment is needed in patients who are diagnosed with incidentalomas. There may be a history of weight gain in patients with Cushing’s syndrome or hypercortisolism. In addition, there would be history of centripetal obesity, easy bruising, hypertension, virilisation and fatigue. In patients with pheochromocytoma, there may be a history of headache, weight loss, anxiety attacks, sweating and cardiac arrhythmias. In aldosteronism the physician has to look for hypertension, fluid retention and history of hypokalaemia [19].
Assessment of Malignant Potential
Evaluation of adrenal incidentaloma has to address two important questions.
(a)
Is it a malignant lesion? If malignant, is it primary adrenal or metastatic?
(b)
Is it a secretory type (functional lesion)?
Being highly vascular, adrenal glands are likely sites for blood-borne metastasis from other organs most notably breast, lung, gastrointestinal tract, kidney, melanoma and lymphoma. Metastatic disease must be suspected in any patient with a history of cancer; three-quarters of incidental adrenal masses prove to be metastatic in patients with such a history. In patients with no prior history of cancer, two thirds of incidentally found adrenal lesions are reported to be benign.
Computed tomography (CT) scanning is effective in separating benign adrenal disease from malignancy. The main indicator of malignancy is size. For example, adrenocortical carcinoma was detected in 2 % of masses <4 cm in diameter, 6 % of masses 4.1–6.0 cm and 25 % of masses >25 cm [20]. Assessment of CT radiographic absorption (CT attenuation coefficient expressed as Hounsfield units, HU) of adrenal masses suggests that a value of >10 HU is an indicator of malignant potential, with specificity of 96–100 % and sensitivity of 68–79 % [21, 22]. Other radiographic features such as heterogeneous appearance of the gland, irregular border of the mass and involvement of surrounding structures are sinister. For patients who are unable to undergo a contrast-enhanced CT because of renal insufficiency or allergy to iodinated contrast media, chemical shift MRI can be performed as CT and MRI demonstrate similar accuracy in the diagnosis of adrenal masses [13].
The finding of intracellular lipid in a solid adrenal lesion by magnetic resonance imaging (MRI) is suggestive of an adrenocortical adenoma. Pheochromocytomas are characterised by hypointensity on T1-weighted MRI and hyperintensity on T2-weighted MRI. The presence of fat in the lesion may suggest a myelolipoma, which are benign non-functioning neoplasms consisting of mature adipose cells and haemopoetic tissue.
Assessment of Functional Status
The main concern is that an adrenal incidentaloma is secreting cortisol. In this case, adrenalectomy may be more hazardous. Firstly, the contralateral adrenal may be suppressed, and a hypoadrenal crisis and hypotension may occur when the adenoma is removed and the circulating cortisol falls to low levels. Secondly, complications of infection, poor wound healing and haemorrhage are more likely if the high pre-operative cortisol levels are not detected and treated prior to operation. An elevated serum cortisol level and suppressed serum adrenocorticotropin (ACTH) or dehydroepiandrostenedione sulphate (DHEAS) are very suggestive of autonomous adrenal cortisol production. However, patients with mild over-secretion may have serum cortisol levels within the normal range and a low dose dexamethasone suppression test is often useful.
Conn’s syndrome should be suspected in any patient with hypertension with or without hypokalaemia. As a screening measure, patients should have an assessment of their electrolytes including bicarbonate and measurement of plasma aldosterone concentration and plasma renin activity with assessment of the ratio.
Clinically silent pheochromocytomas account for 5–11 % of incidentalomas [4, 23, 24]. These frequently present with no specific symptoms and should be excluded in any patient with an incidentaloma. Twenty-four hour urinary free catecholamines provide a useful screening tool with 96 % sensitivity and are widely available [25]. Other tests include plasma or urinary metanephrines and fractionated catecholamines, with sensitivity and specificity of around 95 %. Serum DHEAS as levels may be increased in adrenocortical carcinoma. Serum 17-hydroxyprogesterone should be assessed in view of the possibility of congenital adrenal hyperplasia.
Management
Adrenalectomy is recommended for masses greater than 6 cm in diameter. Homogeneous lesions less than 4 cm diameter are considered low risk and may be followed by scanning. Masses, which measure 4–6 cm, or with heterogeneity, may be followed up or excised, although if features of rapid growth or decreased lipid content are present, surgery would be advisable. Over a 10-year follow-up, less than 30 % increase in size and less than 20 % develop biochemical abnormalities [20].
Tumours of the Adrenal Cortex
Benign (Adenomas)
Functioning Adrenocortical Adenomas
Adrenal Cushing’s syndrome accounts for approximately 10 % all cases of Cushing’s syndrome. The most common cause is a unilateral benign adenoma. In severe, untreated cases it is associated with 50 % mortality at 5 years [26], whilst less clinically obvious cases have a significantly increased morbidity and mortality due to hypertension and secondary diabetes mellitus. Clinical manifestations vary according to the degree of levels of cortisol.
In adrenal Cushing’s, the serum cortisol usually remains high throughout the day and night, or pulses up and down with no relation to time of day. The midnight cortisol in adrenal Cushing’s syndrome is usually very similar to the morning cortisol. A low dose dexamethasone suppression test in adrenal Cushing’s syndrome usually shows no suppression of the serum cortisol. The high circulating cortisol levels suppress the hypothalamo-pituitary axis, resulting in undetectable serum ACTH levels and often mild reductions in LH and TSH levels.
The mainstay of treatment for adrenal Cushing’s syndrome is unilateral adrenalectomy. Serum cortisol levels can be lowered with metyrapone for at least 6 weeks before surgery. Ketoconazole may also be used for this purpose but has a slower time of onset and off-set. The anaesthetic agent etomidate provides an effective short-term method of controlling very severe hypercortisolaemia [27]. Hydrocortisone cover should be administered with the induction of anaesthesia and continued during the peri- and post-operative period due to hypothalamo-adrenal axis suppression. One regimen is a hydrocortisone infusion of 2 mg per hour, or 100 mg i.m. every 6 h until the patient is able to swallow tablets again (usually within 8 h). The oral hydrocortisone is rapidly tapered to a physiological replacement dose of 10 mg on waking, 5 mg at lunch and 5 mg at 1,700-h.
The patient is readmitted about 3 months after surgery to withdraw hydrocortisone and to check the recovery of the hypothalamo-pituitary axis and endogenous cortisol from the contralateral adrenal gland. If the patient is still dependent on hydrocortisone, reassessments are done every 6 months. Most patients recover the function of the contralateral adrenal by about 18 months after surgery, but a small number continue to need hydrocortisone replacement for the long-term. Adrenal adenomas treated successfully by surgery have a good prognosis with a low risk of recurrence.
Conn’s Syndrome (Primary Hyperaldosteronism)
Aldosterone is the main mineralocorticoid produced by the zona glomerulosa; the outer portion of the adrenal cortex. Hyperaldosteronism most commonly occurs as a result of a unilateral adenoma or idiopathic hyperaldosteronism (also known as bilateral adrenal hyperplasia). Rare causes include glucocorticoids-suppressible hyperaldosteronism and aldosterone-producing adrenocortical carcinoma.
Many anti-hypertensive drugs interfere with the assessment of hyperaldosteronism; but patients may be controlled on calcium-antagonists or α-adrenoceptor blockers during investigation. Diuretics, β-adrenoceptor antagonists, angiotensin-converting enzyme inhibitors and angiotensin-2 receptor antagonists should be withdrawn for at least 2 weeks prior to investigations. Initial simple investigations include measurement of serum potassium and bicarbonate and urinary potassium; the presence of a hypokalaemic alkalosis and raised urinary potassium of >30 mmol/24 h is highly suggestive of hyperaldosteronism.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


