Tubular Potassium Transport
David B. Mount
TUBULAR POTASSIUM TRANSPORT
Potassium (K+) functions in a diversity of physiologic processes. Changes in intracellular K+ affect cell volume, intracellular pH, enzymatic function, protein synthesis, DNA synthesis, and apoptosis. Changes in the ratio of intracellular to extracellular K+ affect the cellular resting membrane potential, causing depolarization in hyperkalemia and hyperpolarization in hypokalemia; as a consequence, potassium disorders primarily affect excitable tissues, chiefly heart and muscle. Hypokalemia and hyperkalemia also have a variety of renal and cardiovascular consequences. A large body of experimental and epidemiologic evidence thus implicates hypokalemia or reduced K+ intake in the pathogenesis of hypertension, heart failure, and stroke.1 Hypokalemia also causes a host of structural and functional changes in the kidney, whereas hyperkalemia in turn has a significant effect on the ability to excrete an acid urine due to interference with the urinary excretion of ammonium (NH4+).
Potassium is almost exclusively an intracellular cation, with only 2% of total body K+ contained within the extracellular fluid. Extracellular K+ is maintained within a very narrow range by three major mechanisms. First, the distribution of K+ between the intracellular and extracellular space is determined by the activity of a number of widely expressed and/or ubiquitous transport pathways. A net increase in cellular uptake can thus cause transient hypokalemia, whereas impairment of cellular uptake can lead to hyperkalemia. Second, the colon has the ability to absorb and secrete K+, with significant mechanistic and regulatory similarities to renal K+ transport. However, the colon has a relatively limited capacity for K+ excretion, and a third mechanism, changes in renal K+ excretion, plays the dominant role in responding to changes in K+ intake. Regulated increases in K+ secretion by the connecting tubule (CNT) and the cortical collecting duct (CCD) play a critical role in the response to hyperkalemia and K+ loading, whereas inhibition of K+ secretion and increases in the reabsorption of K+ by the CCD and the outer medullary collecting duct (OMCD) function in the response to hypokalemia or K+ deprivation.
This chapter reviews the renal and extrarenal mechanisms of K+ homeostasis, with primary emphasis on the physiology of renal K+ transport.
Extrarenal Potassium Homeostasis
The intracellular accumulation of K+ against its electrochemical gradient is an energy-consuming process, mediated by the ubiquitous Na+/K+-ATPase. The Na+/K+-ATPase functions as an electrogenic pump, with a transport stoichiometry of three intracellular Na+ ions to two extracellular K+ ions. The enzyme complex is made up of a tissue-specific combination of multiple α,β, and ρ subunits, which are further subject to tissue-specific patterns of regulation. Cardiac glycosides (i.e., digoxin and ouabain) bind to the α subunits of Na+/K+-ATPase at an exposed extracellular hairpin loop that also contains the major binding sites for extracellular K+.2 The binding of digoxin and K+ to the Na+/K+-ATPase complex is thus mutually antagonistic, explaining in part the potentiation of digoxin toxicity by hypokalemia.3 Although the four α subunits have equivalent affinity for ouabain, they differ significantly in the intrinsic K+/ouabain antagonism.4 Ouabain binding to isozymes containing the ubiquitous α1 subunit is relatively insensitive to K+ concentrations within the physiologic range, such that this isozyme is protected from digoxin under conditions wherein cardiac α2 and α3 subunits, the therapeutic targets of digoxin, are inhibited.4 Notably, the digoxin/ouabain binding site of α subunits is highly conserved, suggesting a potential role in the physiologic response to endogenous ouabain/digoxinlike compounds. Consistent with this hypothesis, a mouse strain that expresses α2 subunits with engineered resistance to ouabain is strikingly resistant to ouabain-induced hypertension and to adrenocorticotrophic hormone-dependent hypertension,5 the latter of which is known to involve an increase in circulating ouabainlike glycosides.
Potassium can also accumulate in cells by coupling to the gradient for Na+ entry, entering via the electroneutral Na+-K+-2Cl– cotransporters NKCC1 and NKCC2. The NKCC2 protein is found only at the apical membrane of
the thick ascending limb (TAL) and the macula densa cells, where it functions in transepithelial salt transport and tubular regulation of renin release (see Potassium Transport in the Thick Ascending Limb).6 NKCC1 is widely expressed in multiple tissues,6 including muscle, where it modulates extrarenal K+ homeostasis.7 The cotransport of K+-Cl– by the four K+-Cl– cotransporters (KCC1 to 4) can also function in the transfer of K+ across membranes; although the KCCs typically function as efflux pathways, they can mediate influx when extracellular K+ increases.6
the thick ascending limb (TAL) and the macula densa cells, where it functions in transepithelial salt transport and tubular regulation of renin release (see Potassium Transport in the Thick Ascending Limb).6 NKCC1 is widely expressed in multiple tissues,6 including muscle, where it modulates extrarenal K+ homeostasis.7 The cotransport of K+-Cl– by the four K+-Cl– cotransporters (KCC1 to 4) can also function in the transfer of K+ across membranes; although the KCCs typically function as efflux pathways, they can mediate influx when extracellular K+ increases.6
Skeletal muscle contains approximately 75% of the body’s potassium and exerts considerable influence on extracellular K+. Skeletal muscle Na+/K+-ATPase activity in particular is a major determinant of the capacity for extrarenal K+ homeostasis. Hypokalemia induces a marked decrease in muscle K+ content and Na+/K+-ATPase activity, an altruistic8 mechanism to regulate plasma K+. This adaptation is primarily mediated by dramatic decreases in the protein abundance of the α-2 subunit of Na+/K+-ATPase. In contrast, hyperkalemia due to potassium loading is associated with adaptive increases in muscle K+ content and Na+/K+-ATPase activity.9 These interactions are reflected in the relationship between physical activity and the ability to regulate extracellular K+ during exercise10; exercise training is associated with increases in muscle Na+/K+-ATPase concentration and activity, with reduced interstitial K+ in trained muscles and an enhanced recovery of plasma K+ after defined amounts of exercise.10
Several hormones have been implicated in the control of extrarenal K+ homeostasis. In particular, increases in plasma K+ have a stimulatory effect on insulin levels,11 which in turn stimulates the uptake of K+ by muscles, the liver, and other tissues. In contrast, insulin-stimulated K+ uptake is rapidly reduced by 2 days of K+ depletion, prior to a modest drop in plasma K+,12 and in the absence of a change in plasma K+ in rats subject to a lesser K+ restriction for 14 days.13 Insulin activates muscle Na+/K+-ATPase, inducing the translocation of the Na+/K+-ATPase ct-2 subunit to the plasma membrane with a lesser effect on the al subunit.14 This translocation is dependent on the activity of phosphoinositide-3 (PI-3) kinase,14 which itself also binds to a proline-rich motif in the N-terminus of the α subunit; the activation of PI3-kinase by insulin induces phosphatase enzymes to dephosphorylate a specific serine residue adjacent to the PI3-kinase binding domain. Trafficking of Na+/K+-ATPase to the cell surface also requires phosphorylation of an adjacent tyrosine residue, perhaps catalyzed by the tyrosine kinase activity of the insulin receptor itself.15 In addition, the serum and glucocorticoid-induced kinase 1 (SGK1) plays a critical role in insulin-stimulated K+ uptake via stimulatory effects on Na+/K+-ATPase activity and/or the Na+-K+-2CG cotransport.16 The hypokalemic effect of insulin plus glucose is thus blunted in SGK1 knockout mice, with a marked reduction in hepatic insulin-stimulated K+ uptake.16
The sympathetic nervous system also affects the balance between extracellular and intracellular K+. The uptake of K+ by the liver and muscles is stimulated via β2 receptors. This hypokalemic effect of catecholamines is independent of changes in circulating insulin and has been reported in nephrectomized animals.17 The cellular mechanisms whereby catecholamines induce K+ uptake in muscles include an activation of the Na+/K+-ATPase, likely via increases in cyclicAMP (cAMP).18 Skeletal muscle β-adrenergic receptors also activate NKCC1, which may account for as much as one third of the uptake response.7 In contrast to β-adrenergic stimulation, α-adrenergic agonists impair the ability to buffer increases in K+ induced via intravenous loading or by exercise19; the transport mechanisms whereby this occurs are not known. β-adrenergic stimulation increases K+ uptake during exercise to lessen exercise-induced hyperkalemia, whereas α-adrenergic mechanisms help blunt the ensuing postexercise nadir.19
The efflux of K+ out of cells is primarily mediated by K+ channels, which comprise the largest group of ion channels in the human genome. There are three major subclasses of mammalian K+ channels: the six-transmembrane domain (TMD) family, which encompasses both the voltage-sensitive and Ca2+-activated K+ channels; the two-pore, four TMD family; and the two TMD family of inward rectifying K+ (Kir) channels. There is tremendous genomic variety in human K+ channels. Further complexity is generated by the presence of multiple accessory subunits and alternative patterns of mRNA splicing. Not surprisingly, an increasing number and variety of K+ channels have been implicated in the control of K+ homeostasis and the membrane potential of excitable cells such as muscle and heart, with important, evolving roles in the pathophysiology of potassium disorders. Adrenal K+ channels have also been implicated in the adrenal release of aldosterone induced by hyperkalemia.20,21 The emphasis in this chapter is on renal K+ channels, particularly those in the distal nephron that mediate K+ secretion.
Potassium Transport in the Proximal Tubule
The proximal tubule reabsorbs 50% to 70% of filtered K+ (Fig. 6.1). Proximal tubules generate minimal transepithelial K+ gradients, and the fractional reabsorption of K+ by this nephron segment is similar to that of Na+.22 K+ absorption thus follows that of fluid, Na+, and other solutes,23 such that proximal tubules do not play a direct role in regulated renal K+ excretion. However, changes in Na+-Cl– reabsorption by the proximal tubule have considerable effects on distal tubular flow and distal tubular Na+ delivery, with attendant effects on the excretory capacity for K+. In addition, K+ loading has significant effects on proximal tubular Na+-Cl– reabsorption. Thus, the intravenous infusion of K+-Cl– or increases in the peritubular K+ concentration causes an inhibition of proximal tubular Na+-Cl– reabsorption24; this serves to increase both distal tubular flow and distal tubular Na+ delivery, thus increasing distal K+ secretion after K+ loading.
The mechanisms involved in transepithelial K+ transport by the proximal tubule are not completely clear, although
active transport does not appear to play a major role.23,25 Luminal barium has modest effects on transepithelial K+ transport, suggesting a component of transcellular transport via barium-sensitive K+ channels.26 However, the bulk of K+ transport is thought to occur via the paracellular pathway,26,27 driven by the lumen-positive potential difference in the mid-to-late proximal tubule. The total K+ permeability of the proximal tubule is thus quite high due to the high permeability of the paracellular pathway26,27 The combination of luminal K+ concentrations that are ˜ 10% higher than that of plasma, a lumen-positive potential difference of ˜2 mV, and high paracellular permeability leads to considerable paracellular absorption in the proximal tubule. The tight junction protein claudin-2 plays a key role in paracellular cation transport by the proximal tubule; targeted deletion in knockout mice generates a “tight” epithelium in the proximal tubule, with a reduction in Na+, Cl–, and fluid absorption.28 The loss of claudin-2 expression does not affect the ultrastructure of tight junctions, but does lead to a reduction in paracellular cation permeability.28
active transport does not appear to play a major role.23,25 Luminal barium has modest effects on transepithelial K+ transport, suggesting a component of transcellular transport via barium-sensitive K+ channels.26 However, the bulk of K+ transport is thought to occur via the paracellular pathway,26,27 driven by the lumen-positive potential difference in the mid-to-late proximal tubule. The total K+ permeability of the proximal tubule is thus quite high due to the high permeability of the paracellular pathway26,27 The combination of luminal K+ concentrations that are ˜ 10% higher than that of plasma, a lumen-positive potential difference of ˜2 mV, and high paracellular permeability leads to considerable paracellular absorption in the proximal tubule. The tight junction protein claudin-2 plays a key role in paracellular cation transport by the proximal tubule; targeted deletion in knockout mice generates a “tight” epithelium in the proximal tubule, with a reduction in Na+, Cl–, and fluid absorption.28 The loss of claudin-2 expression does not affect the ultrastructure of tight junctions, but does lead to a reduction in paracellular cation permeability.28
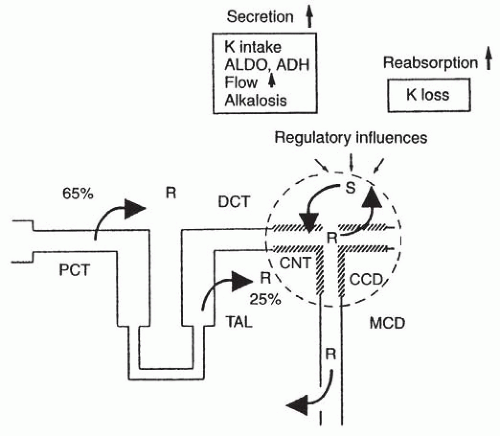 FIGURE 6.1 Potassium transport along the nephron. Approximately 90% of filtered K+ is reabsorbed by the proximal tubule and the loop of Henle. K+ is secreted in the connecting tubule and the cortical collecting duct; net reabsorption occurs in response to K+ depletion, primarily within the medullary collecting duct. PCT, proximal tubule; TAL, thick ascending limb; DCT, distal convoluted tubule; CNT, connecting tubule; CCD, cortical collecting duct; S, secretion; R, reabsorption; ALDO, aldosterone; ADH, antidiuretic hormone; MCD, medullary collecting duct. |
The absorption of K+ across the paracellular pathway is thought to occur via convective transport—solvent drag due to frictional interactions between water and K+—rather than diffusional transport.29 Notably, however, the primary pathway for water movement in the proximal tubule is quite conclusively transcellular, via water channels in the apical and basolateral membrane. Therefore, the apparent convective transport of K+ would have to constitute pseudosolvent drag, with uncharacterized, coordinating interactions between water traversing the transcellular route and the diffusion of K+ along the claudin-dependent paracellular pathway.29
The Loop of Henle and Medullary Potassium Recycling
Transport by the loop of Henle functions in medullary K+ recycling (Fig. 6.2). A considerable fraction of K+ secreted by the CCD is reabsorbed by the medullary collecting ducts and is then secreted into the late proximal tubule and/or the descending thin limbs of long-looped nephrons.30 In potassium-loaded rats there is thus a doubling of luminal K+ in terminal thin descending limbs, with a sharp drop after the inhibition of CCD K+ secretion by amiloride.31 Enhancement of CCD K+ secretion with the treatment of 1-desamino-8-D-arginine vasopressin (dDAVP) also results in an increase in luminal K+ in descending thin limbs.32 This recycling pathway (i.e., secretion in CCD, absorption in the OMCD and the inner medullary collecting duct (IMCD), secretion in the descending thin limb)
is associated with a marked increase in medullary interstitial K+ concentration. Passive transepithelial K+ absorption by the thin ascending limb and active absorption by the thick ascending limb (TAL)33 also contribute to this increase in interstitial K+ (Fig. 6.2). Specifically, the absorption of K+ by the ascending thin limb, TAL, and OMCD exceeds the secretion by descending thin limbs, thus concentrating K+ within the interstitium.
is associated with a marked increase in medullary interstitial K+ concentration. Passive transepithelial K+ absorption by the thin ascending limb and active absorption by the thick ascending limb (TAL)33 also contribute to this increase in interstitial K+ (Fig. 6.2). Specifically, the absorption of K+ by the ascending thin limb, TAL, and OMCD exceeds the secretion by descending thin limbs, thus concentrating K+ within the interstitium.
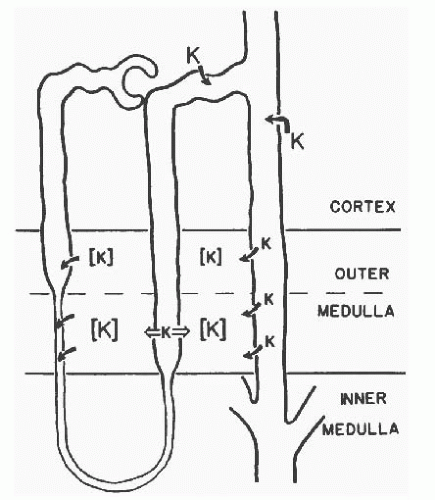 FIGURE 6.2 A schematic representation of medullary K+ recycling. Medullary interstitial K+ increases considerably after dietary K+ loading due to the combined effects of secretion in the cortical collecting duct (CCD), absorption in the outer medullary collecting duct (OMCD), the thick ascending limb (TAL), and the inner medullary collecting duct (IMCD), and secretion in the descending thin limb (see text for details). (From Stokes JB. Consequences of potassium recycling in the renal medulla. Effects of ion transport by the medullary thickascending limb of Henle’s loop. J Clin Invest. 1982;70:219-229.) |
K+ is secreted into descending thin limbs by passive diffusion, driven by the high medullary interstitial K+ concentration. Descending thin limbs thus have a very high K+ permeability, without evidence for active transepithelial K+ transport.34 Transepithelial K+ transport by ascending thin limbs has not been measured; however, as is the case for Na+-Cl– transport,35 the absorption of K+ by thin ascending limbs is presumably passive, via the paracellular pathway.
The physiologic significance of medullary K+ recycling is not completely clear. However, an increase in interstitial K+ from 5 to 25 mM dramatically inhibits Cl– transport by perfused thick ascending limbs.33 By inhibiting Na+-Cl– absorption by the TAL, increases in interstitial K+ would increase Na+ delivery to the CNT and CCD, thus enhancing the lumen-negative potential difference (PD) in these tubules and increasing K+ secretion.33 A high K+ diet also reduces the absorption of K+ by the TAL36 (i.e., there are direct effects on K+ secretion by the TAL). The marked increase in medullary interstitial K+ after dietary K+ loading is also postulated to limit the difference between luminal and peritubular K+ in the collecting duct, thus minimizing passive K+ loss from the collecting duct.
Potassium Transport in the Thick Ascending Limb
Active transepithelial K+ transport across the TAL includes both a transcellular component, via the apical Na+-K+-2Cl– cotransporter NKCC2, and a paracellular pathway (Fig. 6.3). Potassium transport appears to differ in the major morphologic subtypes of TAL cells (i.e., rough- and smooth-surfaced cells). Morphologically, the TAL begins abruptly after the thin ascending limb of long-looped nephrons and after the aquaporin-negative segment of short-limbed nephrons. The TAL then extends into the renal cortex, where it meets its parent glomerulus at the vascular pole; the plaque of cells at this junction forms the macula densa, which functions as the tubular sensor for both tubuloglomerular feedback and the tubular regulation of renin release by the juxtaglomerular apparatus. Cells in the medullary TAL are 7 to 8 µm in height, with extensive invaginations of the basolateral plasma membrane and interdigitations between adjacent cells37; cells in the cortical TAL are considerably shorter, about 2 µm in height at the end of the cortical TAL in rabbits, with less mitochondria and a simpler basolateral membrane.37 Macula densa cells also lack the lateral cell processes and interdigitations that are characteristic of medullary TAL cells.37
Scanning electron microscopy has revealed that the TAL of both rats38 and hamsters39 contains two morpho-logic subtypes, a rough-surfaced cell type (R cells) with prominent apical microvilli and a smooth-surfaced cell type (S cells) with an abundance of subapical vesicles.37,40 In the hamster TAL, cells can also be separated into those with high apical and low basolateral K+ conductance and a low basolateral Cl– conductance (LBC cells), versus a second population with low apical and high basolateral K+ conductance, combined with high basolateral Cl– conductance (HBC) cells.39,41 The relative frequency of the morphologic and functional subtypes in the cortical and medullary TAL suggests that HBC cells correspond to S cells and LBC cells correspond to R cells.39
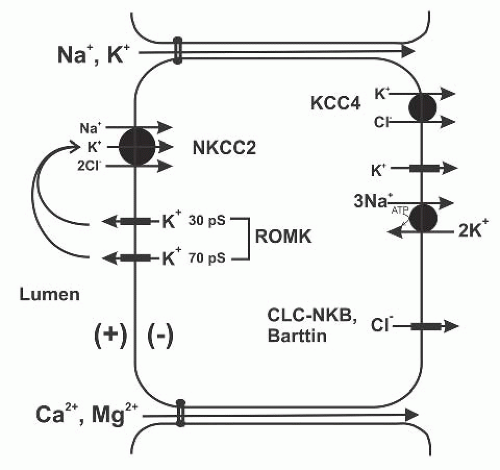 FIGURE 6.3 The potassium transport pathways in the thick ascending limb (TAL); see text for details. NKCC2, Na+-K+-2Cl– cotransporter 2; ROMK, renal outer medullary K+ channel; CLC-NKB, human Cl– channel; Barttin, Cl– channel subunit; KCC4, K+-Cl– cotransporter 4. |
Morphologic and functional heterogeneity notwithstanding, the cells of the medullary TAL, the cortical TAL, and the macula densa are presumed to share the same basic or at least composite transport mechanisms (see Fig. 6.3). Na+-Cl– reabsorption by the TAL is a secondarily active process, driven by the favorable electrochemical gradient for Na+ established by the basolateral Na+/K+-ATPase.42 The Na+, K+, and Cl– ions are cotransported across by the apical membrane by an electroneutral Na+-K+-2Cl– cotransporter; this transporter generally requires the simultaneous presence of all three ions, such that the transport of Na+ and Cl– across the epithelium is mutually codependent and dependent on the luminal presence of K+. An apical Na+-K+-2Cl– cotransport is mediated by the cationchloride cotransporter NKCC2, encoded by the SLC12A1 gene.6 Functional expression of NKCC2 in Xenopus laevis oocytes yields Cl–dependent and Na+-dependent uptake of86Rb+ (a radioactive substitute for K+) and Cl–-dependent and K+-dependent uptake of 22Na+.6 NKCC2 is sensitive to
micromolar concentrations of furosemide, bumetanide, and other loop diuretics.6
micromolar concentrations of furosemide, bumetanide, and other loop diuretics.6
Immunofluorescence indicates the expression of the NKCC2 protein along the entire length of the TAL.6 In particular, immunoelectron microscopy reveals the expression in both rough (R; see previous) and smooth (S) cells of the TAL (see previous).40 NKCC2 expression in subapical vesicles is particularly prominent in smooth cells,40 suggesting a role for vesicular trafficking in the regulation of NKCC2. NKCC2 is also expressed in macula densa cells,40 which have been shown to possess apical Na+-K+-2Cl– cotransport activity. Both tubuloglomerular feedback (TGF) and renal renin secretion are controlled by NKCC2 in the macula densa; luminal loop diuretics block both TGF and the suppression of renin release by the luminal Cl–.6
Microperfused TALs develop a lumen-positive PD during perfusion with Na+-Cl–.43 This lumen-positive PD plays a critical role in the physiology of the TAL, driving the paracellular transport of Na+, K+, Ca2+, and Mg2+ (see Fig. 6.3). Originally attributed to electrogenic Cl– transport,43 the lumen-positive, transepithelial PD in the TAL is generated by the combination of apical K+ channels and basolateral Cl– channels.42 The conductivity of the apical membrane of TAL cells is predominantly, if not exclusively, K+ selective. Luminal recycling of K+ via Na+-K+-2Cl– cotransport and apical K+ channels, along with basolateral depolarization due to Cl– exit through Cl– channels, results in the lumen-positive transepithelial PD.42
Several lines of evidence indicate that apical K+ channels are required for transepithelial Na+-Cl– transport by the TAL.42 First, the removal of K+ from luminal perfusates results in a marked decrease in Na+-Cl– reabsorption by the TAL, as measured by a short circuit current; the residual Na+-Cl– transport in the absence of luminal K+ is sustained by the exit of K+ via apical K+ channels, because the combination of K+ removal and a luminal K+ channel inhibitor (barium) almost abolishes the short-circuit current.44 Apical K+ channels are thus required for the activity of NKCC2, the apical Na+-K+-2Cl– cotransporter; the low luminal concentration of K+ in this nephron segment would otherwise become limiting for transepithelial Na+-Cl– transport. Second, the net transport of K+ across perfused TAL is <10% that of Na+ and Cl–33; -90% of the K+ transported by NKCC2 is recycled across the apical membrane via K+ channels, resulting in minimal net K+ absorption by the TAL.42 Third, the intracellular K+ activity of perfused TAL cells is ˜ 15 to 20 mV above equilibrium, due to furosemide-sensitive entry of K+ via NKCC2.45 Given an estimated apical K+ conductivity of ˜12 m per square centimeter, this intracellular K+ activity yields a calculated K+ current of ˜200 µA per square centimeter; this corresponds quantitatively to the uptake of K+ by the apical Na+-K+-2Cl– cotransporter. Finally, the observation that Bartter syndrome can be caused by mutations in ROMK (renal outer medullary K+ channel),46 a critical component of apical K+ channels in the TAL (see the following), provides genetic proof for the importance of K+ channels in Na+-Cl– absorption by the TAL. Clearance studies in ROMK-deficient mice also indicate that ˜80% of NKCC2 activity is dependent on expression of the ROMK K+ channel with the TAL.47
Three types of apical K+ channels have been identified in the TAL, a 30 pS channel, a 70 pS channel, and a high-conductance, calcium-activated maxi K+ channel.48,49,50 The higher open probability and greater density of the 30 pS and 70 pS channels, versus the maxi K+ channel, suggest that these are the primary routes for K+ recycling across the apical membrane; the 70 pS channel in turn appears to mediate ˜80% of the apical K+ conductance of TAL cells.51 The low conductance 30 pS channel shares several electrophysiologic and regulatory characteristics with ROMK, the cardinal inward-rectifying K+ channel (KIR 1.1) that was initially cloned from the renal outer medulla.52,53 ROMK protein has been identified at the apical membrane of the medullary TAL, the cortical TAL, and the macula densa.54 Furthermore, the 30 pS channel is also absent from the apical membrane of knockout mice with a homozygous deletion of the gene encoding ROMK.55 Notably, not all cells in the TAL are labeled with anti-ROMK antibodies,54,56 suggesting that ROMK might be absent in the HBC cells with high basolateral Cl– conductance and low apical/high basolateral K+ conductance (see also previous).39,41 HBC cells are thought to correspond to the smooth-surfaced morphologic subtype of TAL cells (S cells)39; however, distribution of the ROMK protein by immunoelectron microscopy has not as yet been published, hence correlation cannot be made as of yet between morphology and ROMK expression.
Alternative splicing and alternative promoter use of the KCNJ1 gene encoding ROMK/KIR1.1 generates three different protein isoforms, differing in the extreme aminoterminal amino acid sequence: ROMK1, ROMK2, and ROMK3.57 Although the functional significance of this variation is not clear, these isoforms are differentially distributed along the nephron. ROMK1 is exclusive to the CNT, CCD, and OMCD; ROMK2 is expressed from the TAL to the CCD; and ROMK3 is expressed in the TAL and the distal convoluted tubule (DCT).57 Apical ROMK channels in the TAL are thus a mixture of ROMK2 and ROMK3 proteins.
ROMK clearly plays a critical role in Na+-Cl– absorption by the TAL, given that loss-of-function mutations in this gene are associated with Bartter syndrome.46 The role of ROMK in Bartter syndrome was initially discordant with the data indicating that the 70 pS K+ channel is the dominant conductance at the apical membrane of TAL cells51; heterologous expression of the ROMK protein in Xenopus oocytes had yielded a channel with a conductance of ˜30 pS, suggesting that the 70 pS channel was distinct from ROMK. This paradox was resolved by the observation that the 70 pS channel is absent from the TAL of ROMK knockout mice, indicating that ROMK proteins form a subunit of the 70 pS channel.58 ROMK activity in the TAL is clearly modulated by association with other proteins. That coassociation with other subunits generates the 70 pS channel and is perfectly compatible with
the known physiology of this channel protein. ROMK thus associates with scaffolding proteins NHERF-1 and NHERF-2 via the C-terminal PDZ-binding motif of ROMK; NHERF-2 is coexpressed with ROMK in the TAL.59 The association of ROMK with NHERFs brings ROMK into closer proximity to the cystic fibrosis transmembrane regulator (CFTR) protein.59 This ROMK-CFTR interaction is in turn required for reconstitution of the native ATP and glibenclamide sensitivity of apical K+ channels in the TAL.60
the known physiology of this channel protein. ROMK thus associates with scaffolding proteins NHERF-1 and NHERF-2 via the C-terminal PDZ-binding motif of ROMK; NHERF-2 is coexpressed with ROMK in the TAL.59 The association of ROMK with NHERFs brings ROMK into closer proximity to the cystic fibrosis transmembrane regulator (CFTR) protein.59 This ROMK-CFTR interaction is in turn required for reconstitution of the native ATP and glibenclamide sensitivity of apical K+ channels in the TAL.60
TAL cells are phenotypically defined by the apical expression of uromodulin or Tamm-Horsfall glycoprotein (THP), a TAL-specific, GPI-linked membrane protein that is shed into the tubular lumen as the most abundant protein in normal human urine. THP interacts with ROMK protein in yeast two-hybrid screens and activates the channel in coexpression experiments.61 THP also exhibits functional interactions with NKCC2, activating the cotransporter in coexpression experiments.62 Membrane trafficking of both ROMK and NKCC2 proteins is affected in THP knockout mice, with greater accumulation of both transport proteins in subapical vesicles. THP knockout mice also exhibit a blunted natriuretic and kaliuretic response to furosemide, which is consistent with a partial deficiency in TAL function.62 Mutations of the THP gene cause the allelic disorders familial juvenile hyperuricemic nephropathy, medullary cystic kidney disease type II, and glomerulocystic kidney disease. All three disorders share reduced expression of THP at the apical membrane with retention in the endoplasmic reticulum. The associated defects in NKCC2 and ROMK may explain the renal salt wasting and hyperuricemia that can be associated with these genetic disorders.61,62
Potassium Secretion by the Distal Convoluted Tubule, Connecting Tubule, and Cortical Collecting Duct
Approximately 90% of filtered K+ is reabsorbed by the proximal tubule and the loop of Henle (Fig. 6.1); the final adjustments in renal K+ excretion occur in the downstream distal nephron. The bulk of regulated secretion occurs in the CNT and CCD, whereas K+ reabsorption primarily occurs in the OMCD (see the following). K+ secretion is initially detectable in the early DCT, wherein cells expressing the thiazidesensitive Na+-Cl– cotransporter (NCC) express ROMK, the apical K+ secretory channel (see the following).54 More recent studies in rat kidneys have localized the protein to both DCT1 and DCT2 segments.56 Classically, the CCD is considered the primary site for distal K+ secretion, largely due to the greater ease with which this segment can be perfused and studied. However, as in Na+ absorption (see below),63,64 the bulk of distal K+ secretion appears to occur prior to the CCD,22 within the CNT.65 In addition to a lesser endowment with absorptive apical epithelial Na+ channels (ENaC) and secretory K+ channels, water absorption in the vasopressinresponsive CCD limits K+ secretion by allowing the concentration of luminal K+ to rise toward equilibrium.66 Water permeability of the CNT is considerably lower than that of the CCD,66 with a significant number of early (CNT1) CNT cells that do not express aquaporin-2 in the absence of vasopressin. Aquaporin-2 expression is also low or undetectable in CNT1 cells on a high K+ diet,56 further optimizing these early CNT segments for K+ secretion.
The apical membrane of CNT cells and principal cells contain prominent Na+ and K+ conductances,63,65,67 without a measurable apical conductance for Cl–.68 The entry of Na+ occurs via the highly selective ENaC, which is sensitive to micromolar concentrations of amiloride (Fig. 6.4). This selective absorption of a positive charge generates a lumen-negative PD, the magnitude of which varies considerably as a function of mineralocorticoid status and other factors. This lumennegative PD serves to drive the following critical processes: (1) K+ secretion via apical K+ channels, (2) paracellular Cl– transport through the adjacent tight junctions, and (3) electrogenic H+ secretion via adjacent type A intercalated cells.
The three ENaC subunits are detectable at the apical membrane of CNT cells and principal cells within the CCD, OMCD, and IMCD. Notably, however, several lines of evidence support the hypothesis that the CNT makes the
dominant contribution to amiloride-sensitive Na+ reabsorption by the distal nephron. First, amiloride-sensitive Na+ currents in the CNT are two- to fourfold higher than in the CCD; the maximal capacity of the CNT for Na+ reabsorption is estimated to be ˜10 times higher than that of the CCD.63 Second, targeted deletion of α-ENaC in the collecting duct abolishes amiloride-sensitive currents in CCD principal cells, but does not affect Na+ or K+ homeostasis. Thus, the residual ENaC expression in the late DCT and CNT of these knockout mice easily compensates for the loss of the channel in CCD.69 In contrast, the more extensive deletion of α-ENaC in all aquaporin-2-positive cells in both CNT and CCD causes a profound impairment in Na+ and K+ homeostasis.64 Third, Na+/K+-ATPase activity in the CCD is considerably less than that of the DCT70; this speaks to a greater capacity for transepithelial Na+-Cl– absorption by the DCT and CNT. Fourth, the apical recruitment of ENaC subunits in response to dietary Na+ restriction begins in the CNT, with progressive recruitment of subunits in the downstream CCD at lower levels of dietary Na+71; under conditions of high Na+-Cl– and low K+ intake, the bulk of aldosteronestimulated Na+ transport likely occurs prior to the entry of tubular fluid into the CCD.
dominant contribution to amiloride-sensitive Na+ reabsorption by the distal nephron. First, amiloride-sensitive Na+ currents in the CNT are two- to fourfold higher than in the CCD; the maximal capacity of the CNT for Na+ reabsorption is estimated to be ˜10 times higher than that of the CCD.63 Second, targeted deletion of α-ENaC in the collecting duct abolishes amiloride-sensitive currents in CCD principal cells, but does not affect Na+ or K+ homeostasis. Thus, the residual ENaC expression in the late DCT and CNT of these knockout mice easily compensates for the loss of the channel in CCD.69 In contrast, the more extensive deletion of α-ENaC in all aquaporin-2-positive cells in both CNT and CCD causes a profound impairment in Na+ and K+ homeostasis.64 Third, Na+/K+-ATPase activity in the CCD is considerably less than that of the DCT70; this speaks to a greater capacity for transepithelial Na+-Cl– absorption by the DCT and CNT. Fourth, the apical recruitment of ENaC subunits in response to dietary Na+ restriction begins in the CNT, with progressive recruitment of subunits in the downstream CCD at lower levels of dietary Na+71; under conditions of high Na+-Cl– and low K+ intake, the bulk of aldosteronestimulated Na+ transport likely occurs prior to the entry of tubular fluid into the CCD.
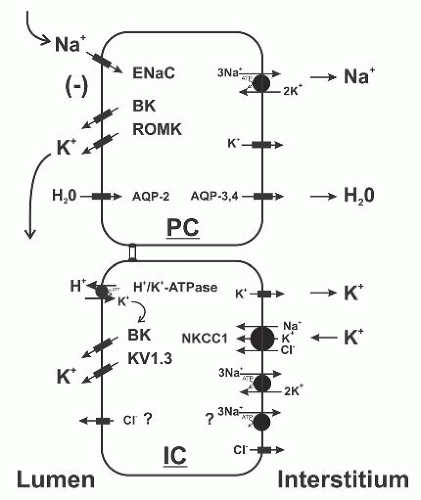 FIGURE 6.4 The transport pathways for potassium in principal/connecting tubule (CNT) cells and intercalated cells. Potassium secretion occurs in CNT and principal cells, driven by the lumennegative potential difference generated by the epithelial Na+ channel (ENaC). In intercalated cells, potassium reabsorbed by apical H+/K+-ATPase recycles across the apical membrane via K+ channels; alternatively, in the setting of hypokalemia or potassium deprivation, it exits the cell via basolateral K+ channels. Potassium that is secreted across intercalated cells enters at the basolateral membrane via NKCC1 and exits via apical BK and Kv1.3 K+ channels. BK, BKK+ channel; ROMK, renal outer medullary K+ channel; Kv1.3, Kv1.3 K+ channel; NKCC1,Na+-K+-2Cl– cotransporter1. |
In principal cells and CNT cells, the lumen-negative potential difference generated by Na+ entry via ENaC drives passive K+ exit through apical K+ channels. Although there is evolving evidence for ENaC— and Na+-independent secretion (see also the following),72 distal K+ secretion is critically dependent on delivery of adequate luminal Na+ to the CNT and CCD,73 essentially ceasing when luminal Na+ drops below 8 mmol/L.74 Dietary Na+ intake also influences K+ excretion, such that excretion is enhanced by excess Na+ intake and reduced by Na+ restriction.73 Secreted K+ enters principal cells via the basolateral Na+/K+-ATPase, which also generates the gradient that drives apical Na+ entry via ENaC (Fig. 6.4).
Two major subtypes of apical K+ channels function in secretion by the CNT and CCD, +/-DCT; a small-conductance (SK) 30 pS channel55,65 and a large-conductance, Ca2+ activated 150 pS (“maxi-K” or BK) channel.65,75 The density and high open probability of the SK channel indicates that this pathway alone is sufficient to mediate the bulk of K+ secretion in the CCD under baseline conditions,76 hence its designation as the “secretory” K+ channel. Notably, SK channel density is considerably higher in the CNT than in the CCD,65 consistent with the greater capacity for Na+ absorption63 and K+ secretion in the CNT. The characteristics of the SK channel are similar to those of the ROMK K+ channel,53 and ROMK protein has been localized at the apical membrane of principal cells.54 SK channel activity is absent from apical membranes of the CCD in homozygous ROMK knockout mice, definitive proof that ROMK is the SK channel.55 The observation that these knockout mice are normokalemic with an increased excretion of K+ illustrates the considerable redundancy in distal K+ secretory pathways55; distal K+ secretion in these mice is mediated by apical BK channels (see the following).77 Loss-of-function mutations in human KCNJ1 are associated with Bartter syndrome; ROMK expression is critical for the 30 pS and 70 pS channels that generate the lumen-positive PD in the TAL (see Fig. 6.3).55,58 These patients typically have slightly higher serum K+ than the other genetic forms of Bartter syndrome,46 and affected patients with severe neonatal hyperkalemia have also been described; this neonatal hyperkalemia is presumably the result of a transient developmental deficit in apical BK channel activity (see the following).
The apical Ca2+-activated BK channel plays a critical role in flow-dependent K+ secretion by the CNT and CCD.75 BK channels have a heteromeric structure, with α-subunits that form the ion channel pore and modulatory β-subunits that affect the biophysical, regulatory, and pharmacologic characteristics of the channel complex.75 BK α-subunit transcripts are expressed in multiple nephron segments, and channel protein is detectable at the apical membrane of principal and intercalated cells in the CCD and CNT.75 The β subunits are differentially expressed within the distal nephron. Thus β1 subunits are restricted to the CNT,75 with no expression in intercalated cells,78 whereas β4 subunits are detectable at the apical membranes of TAL, DCT, and intercalated cells.78 Increased distal flow has a well-established stimulatory effect on K+ secretion, due in part to both the enhanced delivery and absorption of Na+ and to the increased removal of secreted K+.73 The pharmacology of flow-dependent K+ secretion in the CCD is consistent with BK channels,79 and flow-dependent K+ secretion is reduced in mice with targeted deletion of the α1 and β1 subunits.75,80,81 Both mice strains develop hyperaldosteronism that is exacerbated by high-K+ diet,81 leading to hypertension in the α1 subunit knockout.81
One enigma has been the greater density of BK channels in intercalated cells, in both the CCD82 and the CNT.83 This has suggested a major role for intercalated cells in K+ secretion; however, the much lower density of Na+/K+-ATPase activity in intercalated cells has been considered inadequate to support K+ secretion across the apical membrane.84 More recent evidence reveals a major role for the basolateral Na+-K+-2Cl– cotransporter NKCC1 in K+ secretion mediated by apical BK channels (see also Fig. 6.4)85; NKCC1 is expressed almost exclusively at the basolateral membrane of intercalated cells,86 providing an alternative entry pathway for basolateral K+ secreted at the apical membrane. This still begs the question of how basolateral Na+ recycles across the basolateral membrane, in the absence of significant Na+/K+ ATPase activity; one possibility is an alternative basolateral Na+ pump, the ouabain-insensitive furosemide-sensitive Na+-ATPase, a transport activity that has been detected in cell culture models of intercalated cells.85 At the apical membrane, BK-mediated K+ secretion is only partially dependent on luminal Na+87; K+ secretion would eventually hyperpolarize the membrane in the absence of apical Na+ entry, which is mediated by ENaC in principal cells. An intriguing possibility is that apical Cl– channels88 allow for a parallel secretion of K+ and Cl– in intercalated cells (see Fig. 6.4).
BK channels also play a critical role in cell volume regulation by intercalated cells, with indirect, flow-mediated influences on distal K+ secretion. MDCK-C11 cells have an intercalated cell phenotype and express BK α and β4 subunits, as do intercalated cells78; shear stress activates BK channels in these cells, leading to a loss of K+ and cell shrinkage.89 Mice with a targeted deletion of the β4 subunit exhibit normal K+ excretion on a normal diet.84 However, when fed a high K+ diet, which increases urinary and tubular flow rates and tubular shear stress, the β4-knockout mice develop hyperkalemia with a blunted increase in both K+ excretion and urinary flow rates. Intercalated cells from β4 knockout may fail to significantly decrease cell volume in response to a high K+ diet. Intercalated cells thus function as “speed bumps” that protrude into the lumen of distal tubules; flowactivated BK channels reduce the cell volume of intercalated cells after K+ loading, reducing tubular resistance, increasing tubular flow rates, and increasing distal K+




Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
