Fig. 12.1
Ultrasonography of a patient with unilateral renal tuberculosis showing dilated collecting system (caliectasis) and thinning of the renal parenchyma. From Figueiredo et al. [9]; used with permission
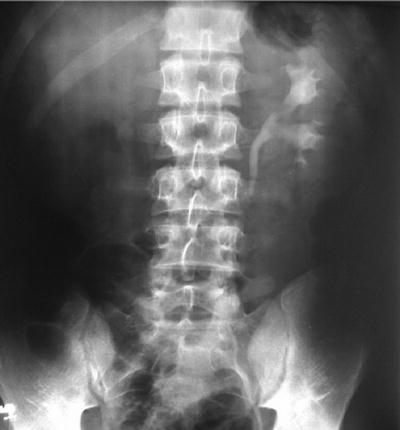
Fig. 12.2
Intravenous pyelogram showing right kidney silence in a patient with renal tuberculosis. From Oliveira et al. [10]; used with permission
Urine and Lower Urinary Tract Examination
Urinalysis may vary from mild changes, such as proteinuria and leukocyturia, to extreme pyuria, sometimes accompanied by hematuria. Urine cultures are regularly negative, unless there is severe bladder dysfunction. However, urine culture aimed at mycobacteria—using Lowenstein-Jensen solid culture medium—may be useful. Multiple samplings should be obtained in order to increase test sensitivity. Mycobacterium culture and identification results provide a specific diagnosis, yet may not be available for 2–3 weeks or longer [6]. Polymerase chain reaction (PER) techniques, such as the direct Gen-Probe MAD test, have been lately used. It is a reliable and fast-performing diagnostic test. Cystoscopy with biopsy is particularly recommended as it allows visualizing and sampling bladder lesions—this is possibly the best test to perform [6].
Pathophysiology
Urogenital tuberculosis is always secondary to a respiratory inoculation, which may be clinically unapparent [7]. Bacilli reach the renal cortex by blood or lymphatic dissemination, where they thrive, before extending to the lower urinary tract. The spreading lesions, most often bilateral, reach the pyramids, pelvis, ureters, and bladder—seminal vesicles, epididymis, and testicles may be also involved in advanced situations [6].
Pathology
Typically, the initial lesion to be found in kidney biopsies is a granuloma with an area of central caseous necrosis and tubular-interstitial inflammation (Fig. 12.3) [11, 12]. However, necrosis may not always be present, what might suggest other conditions such as sarcoidosis or granulomatous vasculitis [6]. Finding of acid-fast bacilli (which will be bright red on staining) by Ziehl-Neelsen stain inside the granuloma is clearly suggestive of tuberculosis. Yet, the finding of a diagnostic granuloma in a percutaneous kidney biopsy occurs by chance, as the disease is more often focal [6]. In more advanced disease involving the lower urinary region, cystoscopy and biopsy of bladder lesions allow reaching diagnosis [11, 12].
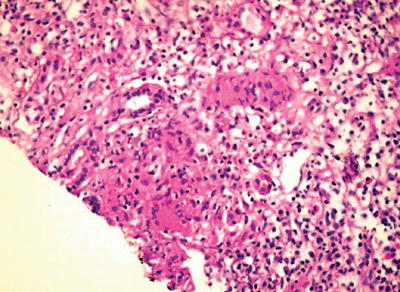
Fig. 12.3
Kidney biopsy of a patient with renal tuberculosis showing severe chronic tubular-interstitial inflammation with tubular atrophy, marked infiltration by lymphoplasmocytes, scattered medium-sized epithelioid cells with noncaseating granulomas, and Langhans giant cells, with one of the granulomas showing caseous necrosis. From Chaudhari et al. [11]; used with permission
Treatment
Urogenital tuberculosis treatment does not diverge from pulmonary tuberculosis therapy. Scarring and development of obstructive lesions may require surgical treatment, besides the placement of endoprosthesis in some special situations. Using a combination of the following drugs, according to the WHO recommendations’—isoniazid, rifampicin, pyrazinamide, streptomycin, ethambutol hydrochloride, and ethionamide—is currently recommended. Therapy usually starts with a combination of isoniazid (300 mg/day), rifampicin (600 mg/day), pyrazinamide (1,600 mg/day), and ethambutol (1,100 mg/day) (“RIPE” schedule), extending for the first 2 months, followed by isoniazid (400 mg/day) and rifampicin (600 mg/day) for the next 4 months. Such doses are suggested for individuals weighting more than 50 kg [13]. Dose adjustments are required for patients with reduced renal function (GFR < 30 ml/min). Many drugs should be administered after a dialysis session and in a thrice-weekly schedule. Antituberculosis drugs for patients presenting with renal failure should undergo the following adjustments: rifampicin, no required adjustment; isoniazid, 75–100 % of the usual dose if GFR is between 10 and 50 mL/min and 50 % if GFR < 10; pyrazinamide, no change in dose, yet in a 24-h interval schedule if GFR is between 10 and 50 mL/min and 48- to 72-h if GFR < 10 mL/min; ethambutol, 50 % of the usual dose if GFR is between 10 and 50 mL/min and 25–50 % if GFR < 10; and streptomycin, no change in dose, yet keeping 24- to 72-h intervals if GFR is between 10 and 50 mL/min and 72–96 h with GFR < 10 mL/min [13].
Antituberculosis therapy interacts with other drugs and increases the risk for hepatotoxicity. Some patients may present with asymptomatic mild elevation of liver enzymes during the first months of treatment, followed by spontaneous decrease. There is usually no need for drug adjustment in those cases. Treatment should only be withdrawn once liver enzymes are over three times the reference values. Following withdrawal, drugs should be reintroduced in the following scheme: rifampicin and ethambutol, followed by isoniazid, and then pyrazinamide. Before reintroducing a drug, liver function tests should be performed. Time for treatment should be counted from the day all drugs have been started again. If reintroducing a drug is not possible, alternative approaches should be attempted. In non-cirrhotic patients, streptomycin (1,000 mg/day) + rifampicin (600 mg/day) + ethambutol (1,100 mg/day) should be given for 2 months, followed by rifampicin (600 mg/day) + ethambutol (1,100 mg/day) for an additional 7-month period. In cirrhotic patients, treatment should be attempted with streptomycin (1,000 mg/day) + ethambutol (1,100 mg/day) + ofloxacin (800 mg/day) for 3 months, followed by ethambutol (1,100 mg/day) + ofloxacin (800 mg/day) for additional 9 months [13].
HIV/AIDS infection in tuberculosis-endemic countries has resulted in a significant increase in pulmonary tuberculosis with negative sputum smears and extrapulmonary infection. Those patients are usually more significantly immunocompromised, more frequently present adverse reactions to antituberculosis drugs, and have higher mortality rates. HIV positivity concomitant with tuberculosis is frequent. Tuberculosis treatment in HIV-infected patients follows the same recommendations as for uninfected individuals. Higher rates of treatment failure and recurrence of tuberculosis have been demonstrated, requiring special attention to the follow-up of such patients. Treatment failure recommendations for the management of recurrence and drug resistant are also similar [13]. No evidence-based, randomized, controlled trials have indicated the best drug schedule for resistant tuberculosis. Recommendations are based on general principles of microbiology and therapy for tuberculosis, observational studies, and specialist opinion. When indication for treatment change emerges, the choice should contemplate an association of the more effective drugs, with the highest probability of cure. An important point in treating patients with resistant tuberculosis is never add one single drug to an already failing regimen, as this may lead to resistance to the added drug. Instead, at least two and preferably three new drugs, to which susceptibility could be inferred, should be combined to lessen the probability of additional resistance. Empirical re-treatment regimens might include a fluoroquinolone; an injectable agent, such as streptomycin (if not used previously and the patient is not from an area of the world having high rates of resistance), amikacin, kanamycin, or capreomycin; and an additional oral agent such as p-aminosalicylic acid (PAS), cycloserine, or ethionamide. Once drug-susceptibility test results are available, the regimen should be adjusted according to the results. Patients having tuberculosis caused by strains of M. tuberculosis resistant to at least isoniazid and rifampicin (multidrug resistant [MDR]) are at high risk for treatment failure and further acquired drug resistance [13].
Tuberculosis is an important opportunistic infection in kidney transplant recipients in endemic areas. Both randomized and nonrandomized studies support the use of isoniazid as prophylaxis in such patients. Clinicians should consider prophylaxis in all kidney transplant recipients in endemic areas or in recipients in non-endemic countries who may be at risk [14].
Leptospirosis
Leptospirosis is a zoonosis caused by organisms of the Leptospira genus, holding worldwide distribution. Its acute evolution, in humans, produces a variety of clinical situations, from nonspecific symptoms to profound jaundice, hemorrhages, meningeal symptoms, and acute kidney injury (AKI). It is a quite uncommon disease in developed countries. Yet, in some areas of endemic leptospirosis, as Thailand and Singapore, it has been associated with AKI in 24 % and 33 % of all cases, respectively [15]. In a 10-year period (1996–2005), 33,174 occurrences were notified in Brazil, and during a single year (2007), 1,547 new cases were notified, mostly in the southern states [13].
Clinical Presentation
Clinical syndromes associated with renal leptospirosis are summarized in Table 12.1. Leptospirosis incubation period varies from 5 to 14 days, with a median time of 10 days [7, 15]. Its clinical presentation varies, depending on the prevalent Leptospira serotype and the geographic area, from a febrile, almost asymptomatic condition, to a severe multisystem disease [7]. Its clinical presentation may occur as (1) a non-jaundice, febrile, auto-limited disease (in 85–90 % of instances); (2) Weil’s syndrome, with jaundice, AKI, hemorrhages, and heart arrhythmias—myocarditis (in 5–10 %); (3) meningitis/encephalitis; and (4) pulmonary hemorrhages, with respiratory insufficiency [16]. It usually follows a two-phase course: the first one (3–7 days) characterized by high fever (100–102 °F), chills, and severe headaches and the second one in which anorexia, nausea, vomiting, diarrhea, and intense myalgia, particularly in lower limbs, dominate. During the first phase it is possible to isolate the Leptospira in blood samples. During the second phase, IgM antibodies appear. Disease severity seems to depend on the intensity of the individual’s humoral immune response [15]. The severest forms of the disease may lead to hemodynamic changes secondary to acute intravascular volume decrease, or a direct toxic effect upon vessel endothelium, and diffuse mounting of capillary permeability [17]. Pulmonary hemorrhagic syndrome may appear independently of other systemic symptoms, sometimes requiring mechanical ventilation, which leads to greater mortality risk [18].
Table 12.1
Clinical syndromes in leptospirosis-associated kidney disease
Clinical presentation | Kidney biopsy |
|---|---|
AKI, fever, jaundice, myalgia, headache, vomiting, dehydration, chills, calf pain, diarrhea, hepatomegaly, anorexia, oliguria, tachypnea, dyspnea, crackles or rhonchi, petechiae, arthralgias, hemoptysis, hematemesis, conjunctival suffusion, edema, obtundation, flapping, constipation, splenomegaly, seizure | Acute tubular necrosis, interstitial nephritis |
Kidney Changes
The kidneys are almost always involved in severe leptospirosis. Non-oliguric leptospirosis-associated AKI is the most frequent presentation, usually unaccompanied by hyperkalemia—hypokalemia may even occur—contrary to AKI associated with other infectious diseases, such as malaria, diphtheria, or meningococcemia. Experimental, as well as clinical, studies have demonstrated that proximal tubule injury and collecting duct vasopressin blunted response may account for such metabolic alterations [19]. Acute, severe jaundice has been linked to functional kidney changes that may encompass falling of glomerular filtration rate and reduced urinary concentration ability [19]. Severe leptospirosis is frequently accompanied by intense jaundice, which may add to the development and severity of the AKI [19]. Rhabdomyolysis and AKI association is well established. Yet, how important rhabdomyolysis may be in leptospirosis-associated AKI is less evident. Increased serum creatine phosphokinase (CPK) levels have been more often noticed in patients with severe leptospirosis-associated AKI than in those with less compromised renal function, suggesting an added risk for AKI from rhabdomyolysis [3]. Proximal tubule damage and collecting duct vasopressin resistance reduce proximal sodium reabsorption and increase free-water clearance, respectively—with resulting polyuria and enhanced natriuria [19]. Increased distal tubule potassium secretion may be induced by increased sodium delivery to distal tubules and raised aldosterone and cortisol levels [19]. Such findings point to a primary proximal tubule defect, with a comparative preservation of distal tubule functional ability to manipulate sodium and potassium. A prospective study on patients with leptospirosis-associated AKI found reduced proximal tubule sodium reabsorption, thus demonstrating the presence of a proximal tubule defect in such patients [20, 21]. Hypokalemia may occur in 45–74 % of patients at admission, potassium replacement being necessary in up to 80 % of instances [19].
Diagnosis
Tests to be used in leptospirosis diagnosis will depend on the disease’s stage the patient is going through. During the initial febrile period, it is possible to visualize the Leptospira by direct examination of blood, to grow it by seeding blood in adequate culture media, or to recover it by laboratory animal inoculation. As it might take several weeks to get a positive result from cultures, usually only a retrospective diagnosis may be obtained in this way [7]. During the second immune phase, Leptospira may be found and grown from urine. Given the difficulties in obtaining a direct diagnosis, serologic tests, such as ELISA IgM macro- and microagglutination tests, have been extensively performed [7].
Pathophysiology
Several factors seem to be involved in the pathophysiology of the kidney lesion, such as a direct nephrotoxic effect by the Leptospira, loss of salt and water, jaundice, and rhabdomyolysis. Experimental studies have suggested that lesions are associated with the physical presence of the organism in the kidney—Leptospira icterohaemorrhagiae has been visualized, as soon as 3–6 h after inoculation, at the mesangium and interstitial tissue [19]. It seems that glomerular capillary Leptospira passage is accompanied by a transitory, moderate, mesangial proliferation [19]. Lack of hyperkalemia is remarkable, even in oligo-anuric individuals, and is a significant characteristic of AKI in such severe infectious disease. It should call the attention of an attending physician, whenever considering differential diagnosis of AKI. Leptospira outer membrane proteins (OMPs) may elicit tubular injury and inflammation through a toll-like receptor (TLR)-dependent pathway, followed by activation of nuclear transcription factor kappa B and mitogen-activated protein kinases and a differential induction of chemokines and cytokines relevant to tubular inflammation [22].
Pathology
Leptospira reach the interstitium by way of peritubular capillaries, causing an acute inflammatory response with focal interstitial edema, lymphocytes, macrophages, plasma cells, and, occasionally, eosinophils infiltrate [19]. Variable degrees of tubular necrosis are always present [19]. Leptospira adhesion to tubule epithelial cells occurs early in the course of the disease, and the infecting organism may be detected even by light microscopy [19]. Importantly, Leptospira antigens’ loading of tubular cells occurs early in disease’s course and may be detected by immunohistochemical staining techniques [23].
Treatment
Quick clinical recovery is the usual outcome—serum creatinine returning to normal levels by the forth to eighth day of symptomatic disease, depending on the severity of kidney involvement. Glomerular filtration rate, proximal sodium reabsorption, fractional potassium excretion, and tubular hydrogen generation complete recovery take place by the third month of follow-up [24]. Yet, a concentration defect may persist for up to 6 months and echoes the severity of AKI [24]. Penicillin seems to reduce symptoms and AKI severity; yet, its advantage has been only demonstrated once started during the first infection week. Early dialysis and treatment of Leptospira-associated AKI seems to be helpful in reducing mortality [25]. Leptospirosis is a disease evolving with low mortality rate. However, in the presence of AKI, mortality rate may be as high as 22 % [19]. Previous studies have examined mortality risk factors associated with leptospirosis—oliguria, old age, episodes of cardiac arrhythmias, and pulmonary involvement are associated with poorer outcome, oliguria being present in over 50 % of reported deaths [19].
Leprosy
Leprosy is a chronic disease associated with infection by Mycobacterium leprae—an obligatory intracellular parasite—an acid-fast bacillus that preferentially infects peripheral nervous system’s Schwann cells and the skin [7]. Its global prevalence has been estimated at 10–15 million, spreading among 106 countries—leprosy world report during the year 2007 included 254,525 new cases [7]. Kidney lesions have been demonstrated in all disease presentations, yet particularly in the multibacillary leprosy form [26–28]. Recent studies show that renal involvement in leprosy is common, with proteinuria in 4.8 % and hematuria in 6.8 % of cases [28]. Risk factors for kidney disease in leprosy include reaction episode, multibacillary classification, and advanced age [28].
Clinical Presentation
Clinical syndromes associated with leprosy are summarized in Table 12.2. Skin and peripheral nervous system damages are leprosy hallmarks. Apparently, host immune response seems to be determinant on the clinical pattern. Two different immunological complications in leprosy course may occur, sporadically intensifying symptoms: (1) a so-called reversal reaction (type 1), a clinical presentation associated with paucibacillary leprosy pattern and (2) “erythema nodosum leprosum” (type 2), frequently associated with multibacillary disease [7]. Leprosy has been classified in four different forms, according to WHO: indeterminate, tuberculoid, dimorphic, and virchowian forms [7]. Diagnosis and classification are dependent upon the clinical presentation and laboratory tests—lesion direct bacilli counting allows classifying leprosy lesions as pauci or multibacillary.
Table 12.2
Clinical syndromes in leprosy-associated kidney disease
Clinical presentation | Kidney biopsy |
|---|---|
Polyarthritis, proteinuria, hematuria, urinary concentration and acidification defects, AKI, CKD | Diffuse proliferative lesion, amyloidosis, acute tubular necrosis, crescentic nephropathy, membranoproliferative nephropathy, membranous nephropathy, mesangial proliferative lesion, interstitial nephritis, glomerular sclerosis |
Kidney Changes
Kidney involvement reports started appearing around 1937, from autopsy studies of patients diagnosed as having died from leprosy [27]. From then on, a series of autopsy and kidney biopsy studies have attempted to elucidate kidney involvement in leprosy. Acute and chronic, nonspecific, glomerular and interstitial lesions—besides amyloid deposits—have been linked to the disease. Daher et al. found kidney changes in 35/923 patients (65 % had multibacillary leprosy) [28]. Risk factors for kidney involvement were old age, having immunological complications and being classified as multibacillary [28]. Glomerular involvement is the more prevalent structural change associated with leprosy, yet with a variable reported prevalence. Kidney biopsy studies on leprosy patients place prevalence at approximately 37 %—higher among multibacillary individuals [29]. Glomerular lesions were strongly associated with occurrence of erythema nodosum, even though lesions have been also reported with no such complication. Almost all known morphological glomerular lesions have been reported, except for focal segmental glomerular sclerosis (FSGS) [30]. Yet membranoproliferative glomerulopathy, so often associated with infectious diseases, has been reported slightly more frequently than other forms [30].
Pathophysiology
Mechanisms leading to leprosy-associated glomerular lesions have been only partly elucidated. Despite bacilli being found in glomerular lesions, no clear evidence for direct Mycobacterium leprae involvement in their genesis exists. Immunological mechanisms may be required: serum complement may be reduced; subendothelial immune complexes have been demonstrated by electron microscopy; IgA mesangial deposition has also been detected. Circulating immune complexes typically accompany erythema nodosum leprosum, with its conceivable deposition in vessels and tissues, including glomeruli. Mycobacterium leprae antigens may be freed, once antibiotics are initiated [31]. Alternatively, antibodies directed toward antigens somewhere inside the glomerulus may complex and deposit locally [31]. However, not every leprosy-associated kidney lesion relates with the concomitant development of erythema nodosum leprosum, thus suggesting glomerular lesions multifactorial origin. Significant reduction on cellular immune response occurs in virchowian leprosy with humoral immune response hyperactivation, which might facilitate immune complex formation and development of glomerular lesions [29].
Tubular dysfunction occurs with some frequency (from 25 % to 85 %), either in multi- or in paucibacillary leprosy [5]. Urine acidification defect appears in 20–32 %, whereas inability to concentrate urine may occur in up to 85 % of leprosy patients [5]. Immunohistochemical examination of kidney samples identified IgM, C3, and, less often, IgA and IgM deposits in the mesangium and capillary basal membranes [31]. Electron microscopy substantiates the presence of mesangial and subendothelial or subepithelial, granular dense deposits [29]. The complement may be reduced in some patients, supporting the idea of an immune-mediated lesion [29].
Urine Changes
Leprosy has been often associated with hematuria, especially in its virchowian form, and with erythema nodosum leprosum, even in the absence of glomerular changes. Microscopic hematuria accompanies virchowian form leprosy in 12–17 % of instances—it often disappears after a couple of months on treatment [29]. Several studies have reported the occurrence of proteinuria (from 2 to 68 %), more frequently associated with immunological complications of multibacillary disease [29]. It usually varies from 0.4 to 8.9 g/24 h, yet nephrotic syndrome is an unusual presentation. Virchowian form of leprosy is more frequently associated with proteinuria and presence of leukocytes, red blood cells, and casts in urine, being uncommon in other forms of the disease [5].
Pathology
Glomerular Lesions
Renal tissue reaction to M. leprae could be induced by various local immunologic or physiological factors. The great variety of lesions suggests a heterogeneous disease, though dependent on a single cause—immune complexes quantity and quality may stand for a divider [5, 31–37]. Adequate kidney biopsy was obtained from 54 cases of leprosy: 45 were lepromatous form, 4 tuberculoid, and 5 belonged to borderline form of leprosy. Membranous nephropathy in 17 (32 %) was the commonest type of glomerular lesion followed by diffuse proliferative lesion in 12 (22 %) and membranoproliferative lesion in 6 (11 %); two samples presented a crescentic nephropathy. Specific glomerular lesions in leprosy include epithelioid granuloma with Hansen’s bacilli in the kidney [32]. Figure 12.4 depicts some features of leprosy kidney disease [32]. Diffuse, endocapillary, proliferative process, with numerous neutrophils occluding peripheral capillary loops, can also be found in leprosy. Figures 12.5 and 12.6 demonstrate such lesions [33]. Electron microscopy may show immune complex-type, electron-dense deposits in the subendothelial area, with electron-dense humps. Figure 12.7 depicts such humps [34]. Crescent formation has also been described in leprosy. Figure 12.8 depicts a leprosy-associated crescent [37].
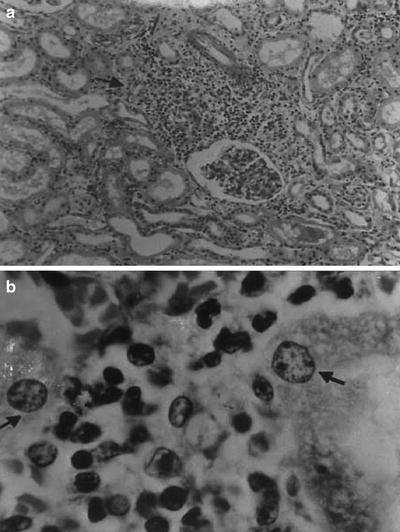
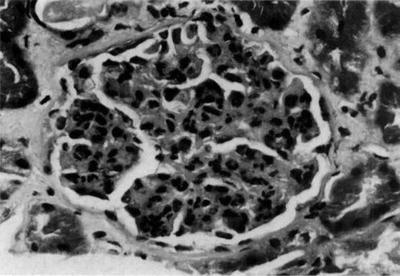
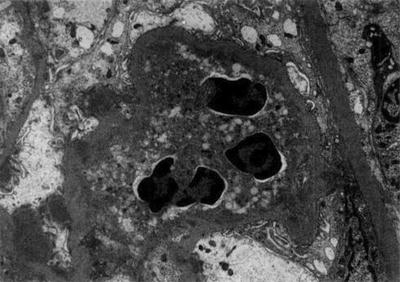
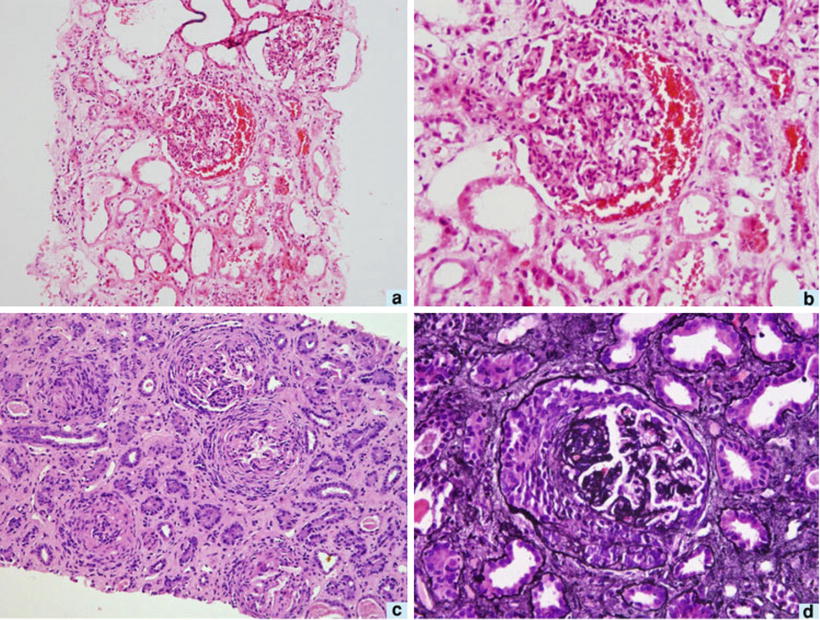
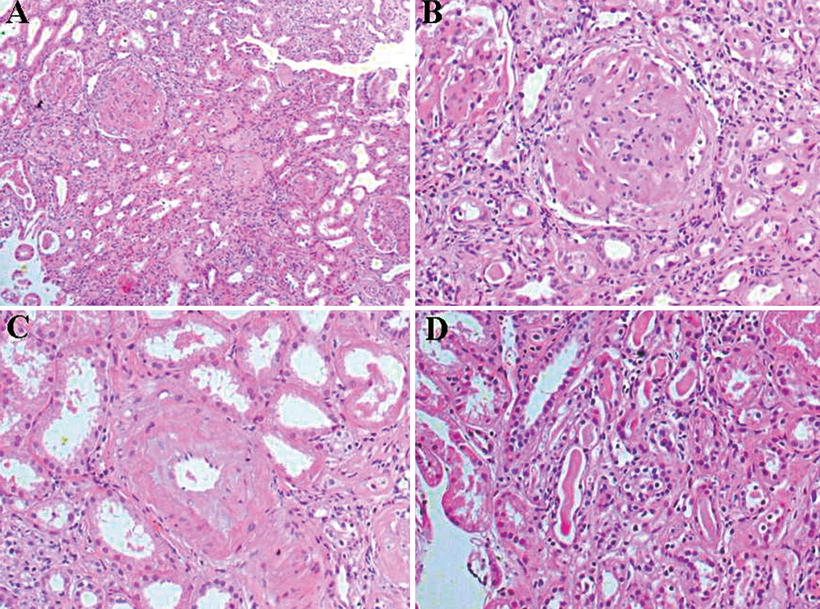
Fig. 12.4
Leprosy-specific renal lesions characterized by the presence of epithelioid granuloma in the renal parenchyma (a), H&E; original magnification ×40; Hansen’s bacilli in the kidney (b); Faraco-Fite stain; original magnification ×400. From Nakayama et al. [32]; used with permission
Fig. 12.5
Glomerulus from a patient with leprosy showing a diffuse, endocapillary, proliferative process with numerous neutrophils occluding the peripheral capillary loops, H&E. From Ahsan et al. [33]; used with permission
Fig. 12.6
Electron photomicrograph of a patient with leprosy showing a neutrophil occluding a peripheral capillary loop. Immune complex-type, electron-dense deposits can be seen in subendothelial areas, whereas electron-dense “humps” are noted in the subepithelial space, consistent with a post-infectious glomerulonephritis. From Ahsan et al. [33]; used with permission
Fig. 12.7
Kidney biopsies from the same patient with leprosy. Composite panel of photomicrographs from first and second biopsies. The first biopsy shows hypercellular glomerulus with hemorrhage in Bowman’s space, interstitial edema, red cell casts in the tubules, and features of acute tubular necrosis (a; H&E, ×100). The glomerulus shows endocapillary cell proliferation with focal neutrophil infiltration (b; H&E, ×200). The second biopsy shows crescent formation in several glomeruli with tubular atrophy and interstitial fibrosis (c; H&E, ×40). Silver-methenamine stain shows a compressed glomerular tuft and overlying cellular crescent (d; ×200). From Sharma et al. [34]; used with permission
Fig. 12.8
Kidney biopsy from a patient with leprosy and chronic kidney disease showing amyloid deposits, H&E, ×200 (a); glomeruli without mesangial proliferation, with amyloid deposit in mesangium, H&E, ×400 (b); amyloid deposit, H&E ×200 (c); tubules without abnormalities, H&E ×200 (d). From Silva Junior et al. [37]; used with permission
Tubulointerstitial Lesions
Interstitial nephritis has been reported chiefly in patients with lepromatous leprosy and seems to relate with long-time illness and extended therapy—such lesion may be the more regular histological finding in leprosy [35].
Chronic Kidney Disease and Leprosy
ESRD has been reported as cause of death in patients with leprosy [36]. ESRD in leprosy has been associated with amyloidosis, more often accompanying virchowian form leprosy. Figure 12.6 depicts amyloid kidney disease in leprosy [33]. Amyloid has been detected in as short an evolution period as 2 years, suggesting that a long disease course may not be necessary for its development [38]. Elevated serum amyloid A levels have been shown during episodes of immunological complications, remaining elevated for several months [38]. In India, where leprosy prevalence is high, almost 50 % of patients have some renal abnormality, yet ESRD has seldom been cause of death [38].
Drug-Associated Renal Changes
Despite renal changes associated with drugs used in leprosy therapy being unusual, acute kidney injury (AKI) described as acute tubular necrosis, acute interstitial nephritis, or papillary necrosis have been reported [28]. Both rifampicin (intermittently, in high doses) and dapsone have been implicated in interstitial nephritis and intravascular hemolysis with AKI [29].
Treatment
Leprosy treatment encompasses specific therapy to overturn M. leprae, avoid immunological complications, and prevent physical deformities, simultaneously promoting physical and psychosocial rehabilitation. Additionally, health authority notification is mandatory [13]. WHO-standardized leprosy therapy includes rifampicin, dapsone, and clofazimine. Prednisone (1–2 mg/kg/day) and nonsteroidal anti-inflammatory drugs (NSAI) may be used to control acute immunological episodes. Erythema nodosum leprosum (ENL) may sometimes have a protracted course (months or years) and is usually treated with NSAIDs, steroids, thalidomide, clofazimine, and pentoxifyline. Management of ENL can be handled with corticosteroids, or corticosteroids + clofazimine. The ideal corticosteroid dose has not been established, but should not exceed 1 mg/kg body weight, during no more than 12 weeks. Addition of clofazimine (100 mg thrice daily, for a maximum of 12 weeks) to corticoid is indicated when severe ENL does not respond to corticosteroid therapy. Analgesics can be used to control fever and pain. Multidrug therapy for leprosy should be continued [13].
It must be kept in mind that all are potentially nephrotoxic. Hemodialysis or kidney transplant are alternatives in treating leprosy ESRD. Posttransplant immunosuppression apparently does not modify leprosy response to drugs. Leprosy is rare in kidney transplant patients, being seldom reported from endemic areas. Treatment is not different from non-transplant patients and immunosuppressive therapy should not be discontinued [39].
Systemic Histoplasmosis
Systemic histoplasmosis is an infectious disease caused by Histoplasma capsulatum—a dimorphic fungus [40, 41]. Disease transmission occurs by fungus inhalation [40]. The illness has worldwide dissemination, having been found in more than 60 countries, in almost every tropical region [40, 41]. It is highly prevalent in the Americas and Africa, being endemic in certain areas of the USA, Argentina, and Brazil. Over 500,000 subclinical infectious instances have been estimated to occur yearly. However, only one among 2,000 or 5,000 instances results in severe clinical histoplasmosis [41].
Clinical Presentation
Histoplasmosis may present a variety of symptoms, ranging from an asymptomatic illness in immunocompetent individuals (90–95 % of instances) to disseminated histoplasmosis affecting several organs or systems [42]. Acute histoplasmosis is a self-limited disease, fever, shivering, headache, and cough being more often present. Disseminated histoplasmosis is more frequently seen in immunocompromised individuals, especially in AIDS patients [42]. A positive intradermal histoplasmin reaction is, many times, the only clue to the diagnosis. In up to one-third of instances, calcified spots in the lungs or in reticuloendothelial organs can be detected. Acute pulmonary, chronic pulmonary, and skin or disseminated histoplasmosis are the more frequent disease presentations, the last one specially affecting immunocompromised individuals [41]. Infants, patients undergoing leukemia or lymphoma therapy, and HIV-positive individuals are particularly vulnerable (22–85 % affected) [40, 41]. Disseminated tuberculosis, leukemia, and lymphoma should be considered for differential diagnosis. Symptoms usually include fever, wasting, diarrhea, vomiting, palpable liver and spleen, augmented lymph nodes, pulmonary infiltrates, and blood changes (anemia, leucopenia, and low platelets count) [40–42]. Skin lesions—papules with necrotic center, covered by bloody crusts—may appear. Other skin lesions may occur, such as petechiae, macules, pruriginous and squamous papules, nodules, pustules, ulcers, and vegetating lesions [41]. In 164 HIV-positive patients with disseminated histoplasmosis, fever (95 %), cough (76 %), wasting (73 %), diarrhea (61 %), weakness (56 %), anorexia (48 %), and vomiting (39 %) were the main complaints [43].
Diagnosis and Laboratory Findings
Histoplasma Identification
Diagnosis of histoplasmosis is backed by the finding of positive antigens or antibodies against the parasite, by direct fungus staining on fluids or tissues or fungus culture [41, 42]. In the acute pulmonary disease presentation, sputum or bronchoalveolar lavage yields the best materials where to look for fungus occurrence. Yet those turn not to be the best diagnostic sources in the chronic disease presentation form—lung biopsy must be usually performed to clarify the clinical picture [41]. Multiple organs are compromised in disseminated histoplasmosis. Peripheral blood, bone marrow aspirate, or some other organ biopsy may be used in search of fungus. H. capsulatum has been isolated in AIDS patients in up to 70 % by blood cultures, using a cell lysing technique (DuPont Isolator) [40, 41]. Additionally, bone marrow cultures have yielded positive results in excess of 60 % [40]. Tzanck’s cell diagnostic test on scuffed material from molluscum-like skin lesions allows the causal agent direct microscopic identification [40]. H. capsulatum is a slow-growing organism: in vitro colonies take up 3 weeks to mature, sometimes up to 2 months [41].
Histoplasmin Skin Test
Histoplasmin is an antigen obtained from cultures of Histoplasma capsulatum that has diagnostic usefulness similar to that of tuberculin when tested by intradermal injection. The histoplasmin skin test has been used in epidemiological surveys performed in endemic areas with apparently good clinical correlation [41].
Other Laboratory Tests
In endemic areas, a significantly increased lactic dehydrogenase (LDH) serum level (usually ≥1,000 UI/l) is highly suggestive of the so-called histoplasmosis endofagocytic syndrome [43]. Abnormally high creatinine, urea, bilirubin, alkaline phosphatase, LDH, AST, and ALT serum levels, accompanied by low protein and albumin serum levels and hematological indexes, have been more often found in AIDS patients with histoplasmosis, compared with those without histoplasmosis—viral loads were higher and CD4 levels lower [43].
Kidney Involvement and Pathophysiology
Previous studies have demonstrated that different fungus species, e.g., Candida, Aspergillus, Cryptococcus, Histoplasma, and some species from the Mucorales genus, may affect the kidney, producing symptoms that may range from minor urine sediment abnormalities to advanced kidney disease and death [44]. Systemic fungus infections have become particularly significant, since the number of surviving immunocompromised patients has substantially expanded. Nephrotic range proteinuria has been demonstrated in HIV-positive patients having disseminated histoplasmosis. Mesangial immune deposits including histoplasma antigens have been shown in kidney biopsies [45]. Antifungal therapy (itraconazol) significantly reduced proteinuria [45].
Histopathology
Granulomatous interstitial nephritis has been occasionally reported. In a case report, by Ahuja et al., an HIV-positive patient with histoplasmosis presented with hematuria, mild proteinuria (365 mg/day), and granular casts. Lymphocytes, monocytes, and plasma cells interstitial infiltrates, non-caseous granulomas, mild interstitial fibrosis and tubular atrophy, and H. capsulatum identification were the main changes on biopsy [46]. Granulomatous interstitial nephritis and glomerular capillary fibrosis, with identified Histoplasma corpuscles, have been reported [47, 48]. Figures 12.9, 12.10, 12.11, 12.12, 12.13, and 12.14 show renal biopsy features in the setting of disseminated histoplasmosis [47–52], and Table 12.3 depicts kidney changes associated with the disease.
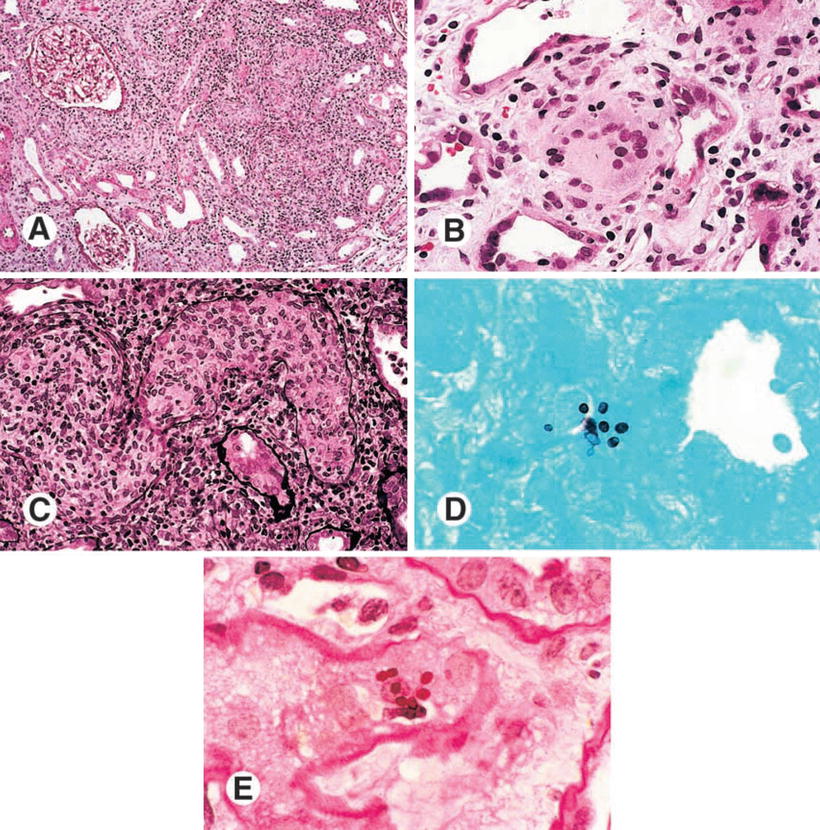
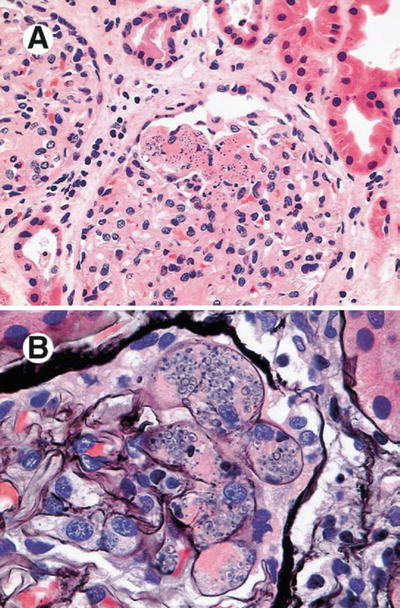
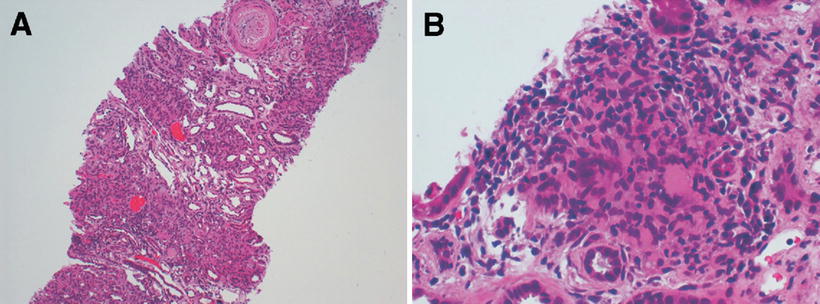
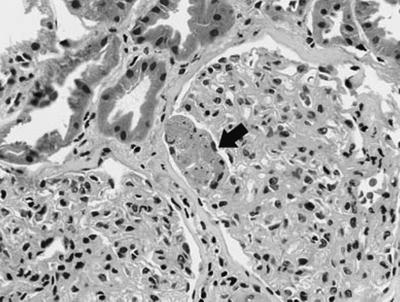
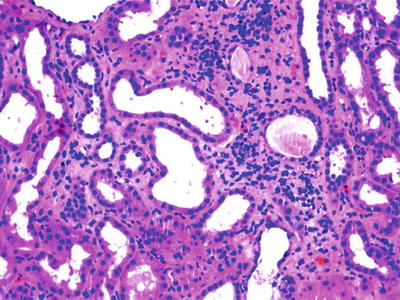
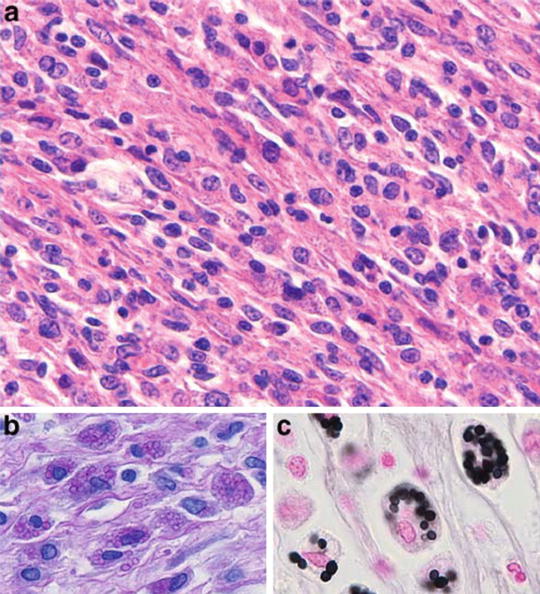
Fig. 12.9
Histopathological findings of a kidney biopsy from a patient with histoplasmosis showing (a) interstitial infiltrate and tubulitis (PAS, ×100), (b) Langhans giant cells in the interstitium (H&E, ×400), (c) obstruction of tubular lumen by a granuloma (Methenamine silver, ×250), (d) ovoid structures with 2–4 mm in the interstitium and tubular epithelium (Giemsa, ×1,000), (e) organisms stained by PAS, forming small groups in the peritubular interstitium (×1,000). From Nasr et al. [47]; used with permission
Fig. 12.10
Renal biopsy of a transplanted patient with histoplasmosis showing fibrosis on glomerular capillary vessels (a, H&E) and microorganisms with characteristics of fungus near glomerular capillaries (b, Silver). From Sethi [48]; used with permission
Fig. 12.11
Renal biopsy of a patient with histoplasmosis showing interstitial infiltrate (a) and occasional giant foreign bodies (b). From Adams and Cook [49]; used with permission
Fig. 12.12
Renal biopsy of a transplanted patient with histoplasmosis showing acute tubular necrosis with microthrombi in the capillaries (arrow). From Dwyre et al. [50]; used with permission
Fig. 12.13
Renal biopsy of a patient with HIV and histoplasmosis showing interstitial inflammatory infiltrate, H&E, ×100. From Bani-Hani et al. [51]; used with permission
Fig. 12.14
Renal biopsy of a patient with idiopathic immunodeficiency showing histiocytic infiltrate, associated with lymphocytes (a), cytoplasm showing granular aspect caused by numerous intracellular organisms compatible with Histoplasma, PAS (b) and Grocott (c). From den Bakker et al. [52]; used with permission
Table 12.3
Clinical syndromes in histoplasmosis-associated kidney disease
Clinical presentation | Kidney biopsy |
|---|---|
Diarrhea, dyspnea, fever, weakness, wasting, nausea, cough, dysphagia, abdominal pain, swollen lymph nodes, nephritic syndrome, edema, AKI
Related posts: General Approach to the Diagnosis and Management of Glomerular Diseases
Childhood Onset Nephrotic Syndrome General Approach to the Diagnosis and Management of Glomerular Diseases
Childhood Onset Nephrotic Syndrome
 Membranoproliferative Glomerulonephritis Membranoproliferative Glomerulonephritis
 Thrombotic Microangiopathies: Thrombus Formation Due to Common or Related Mechanisms? Thrombotic Microangiopathies: Thrombus Formation Due to Common or Related Mechanisms?
 Glomerular Disease in Pregnancy Glomerular Disease in Pregnancy
 Anti-glomerular Basement Membrane Disease Anti-glomerular Basement Membrane Disease
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|