Anti-GBM disease
ANCA-associated vasculitis
• May present with rapidly progressive renal failure and anuria
• Systemic prodromal symptoms are uncommon
• Dialysis dependency at presentation rarely associated with renal recovery
• Pulmonary hemorrhages in 50–70 % cases
• Anti-GBM Ab positive (some serum anti-GBM antibody negative cases reported)
• ANCA positive in 30–47 % cases
• Glomerular crescents of similar age
• Linear immunoglobulin deposition
• May present with rapidly progressive renal failure
• Systemic prodromal symptoms are common, except in cases of renal limited vasculitis
• Dialysis dependency at presentation associated with renal recovery in 40–60 % cases
• Pulmonary hemorrhage in 25 %
• Anti-GBM negative, ANCA positive in 95 % cases
• Glomerular lesions of various ages
• Pauci-immune
Lung hemorrhage may be subclinical and present as an iron deficiency anemia or as breathlessness with cough. In more advanced cases, hemoptysis is a presenting feature, and there may be a drop in hemoglobin which is not explained by any other signs or symptoms. Radiological examination demonstrates diffuse alveolar airspace shadowing, which cannot easily be distinguished from pulmonary edema, although it is said that apical and costophrenic spaces are less often affected (Fig. 9.1). Pulmonary function tests may be useful to confirm hemorrhage through the finding of elevated corrected gas transfer coefficient (KCO). This may also provide a non-radiological means of monitoring resolution of lung hemorrhage. In a recent series from China, patients older than 65 years were reportedly less likely to have hemoptysis and had marginally better presenting creatinine, although they had a greater mortality than their younger counterparts during follow-up [6]. These data confirm findings in earlier series that there was a higher incidence of pulmonary hemorrhage in younger male patients [4].
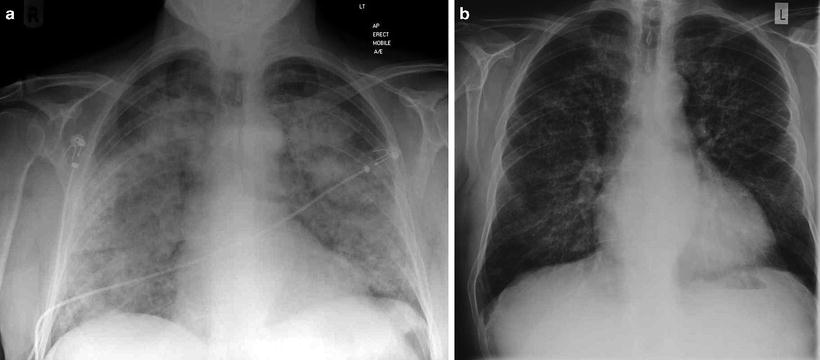
Fig. 9.1
(a) Chest radiograph of a patient who presented acutely with anti-GBM disease demonstrating pulmonary hemorrhage with relative sparing of costophrenic angles and (b) 3 days later following immunosuppression and plasmapheresis
Those patients with double positivity (ANCA and Anti-GBM antibodies) have a renal prognosis similar to that in isolated anti-GBM disease, but their presentation may encompass significant constitutional symptoms and their tendency to relapse is more in keeping with ANCA-associated vasculitis [10, 18–20]. Patients older than 65 were more often double positive for ANCA and anti-GBM antibodies in a large Chinese cohort [6].
Pathology
The characteristic pathologic feature of the disease is a crescentic glomerulonephritis, with the majority of glomeruli showing crescents of a similar age (unlike the pattern found in primary systemic vasculitis; see Table 9.1) (Figs. 9.2 and 9.3). Fibrinoid necrosis may also be present. Furthermore, there may be a florid alveolitis, associated with pulmonary hemorrhage, found in over half the patients (Fig. 9.4). Immunohistochemistry reveals linear immunoglobulin deposition, generally IgG with C3, while IgA and IgM are deposited less commonly (Fig. 9.5). The diagnosis relies on the demonstration of linear immunoglobulin deposition along the GBM in the context of a crescentic glomerulonephritis. Serum anti-GBM antibodies, detected by ELISA, multiplex-based methodology, or Western blotting have high sensitivity and specificity for disease. Rare cases in which circulating anti-GBM antibodies are not detected by conventional means have been described [21]. A number of different ELISA assays and multiplex-based detection systems are available, with most assays demonstrating similar degrees of sensitivity (>95 %) but variable specificity, ranging from 91 to 100 % [22, 23]. These autoantibodies are almost never found using conventional techniques in normal individuals and thus their detection represents a highly sensitive and specific assay for the disease [24]. However, patients with HIV or HCV infection, and polyclonal B cell activation, may be anti-GBM positive without any signs of disease, making the test interpretation more problematic in these scenarios [25]. Overall, since there may be false-positive and false-negative cases of anti-GBM antibody, over-reliance on serum analysis may be misleading and confirmation of the diagnosis necessitates renal biopsy.
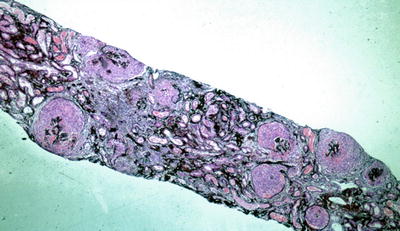
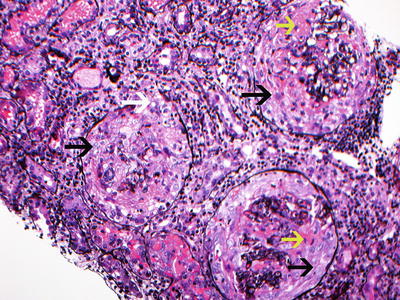
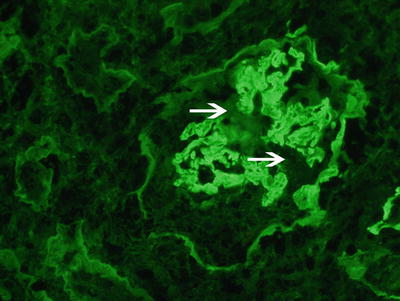
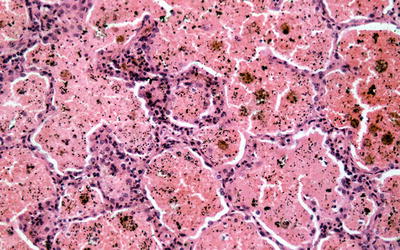
Fig. 9.2
Low power silver methenamine stain of a renal biopsy demonstrating diffuse crescentic glomerulonephritis with cellular crescents of similar age (×100)
Fig. 9.3
Silver stain showing 3 glomeruli with large circumferential cellular crescents with necrosis. Note disruption of Bowman’s capsule. There is prominent interstitial inflammation present. Red blood cells are present in tubules. (black arrow points at cellular crescents, yellow arrow points at fibrinoid necrosis, and white arrow points at disruption of Bowman’s capsule. Jones silver methanamine stain 200x). Courtesy of Dr. Sanjeev Sethi, Mayo Clinic
Fig. 9.4
Immunofluorescence microscopy showing linear staining for anti-IgG along the glomerular capillary walls. Note disruption of the linear staining necrosis/crescent (white arrow points at disruption, 400x). Courtesy Dr. Sanjeev Sethi, Mayo Clinic
Fig. 9.5
Section of lung from a patient with Goodpasture’s disease and alveolar hemorrhage demonstrating hemosiderin-laden macrophages and red blood cells filling the alveolar space
Recently, following methods of purification using autoantigen affinity columns, low titers of predominantly IgG4 and IgG2 anti-GBM antibodies have been found in sera from normal Chinese and Swedish populations [9, 26]. The significance of these “natural” anti-GBM antibodies in healthy individuals is uncertain, although they suggest that healthy individuals have effective methods of regulation, preventing these low-titer antibodies from inducing disease.
Serum ANCA has to be tested for any patient with anti-GBM disease as they may warrant different management strategies with maintenance immunosuppression to prevent relapsing vasculitic symptoms.
Outcome
Both patient and renal prognosis are dependent on the severity of disease at presentation, with the presenting creatinine and extent of glomerular involvement clearly relating to the likelihood of renal recovery [4, 16, 27]. Again, unlike systemic ANCA-associated vasculitis, where 50–60 % of dialysis-dependent patients will recover independent renal function even temporarily [28], in anti-GBM disease dialysis dependency at presentation with 100 % glomerular involvement on biopsy is generally associated with irrecoverable renal failure, even with adequate therapy [4]. Overall, in patients with more severe renal dysfunction at presentation, in particular those requiring dialysis, renal recovery is less likely (Table 9.3 [4, 16, 17, 27, 29]). Other degrees of severity generally respond with some degree of renal recovery following immunosuppression and plasmapheresis (see treatment below) [4, 16, 27].
Risk Factors
Genetic Susceptibility
Human Leukocyte Antigen Genes
The association of anti-GBM disease with HLA DR15 (previously known as HLA-DR2) was first reported over 20 years ago and remains one of the strongest HLA associations for autoimmune disease. Since then a number of studies, including a large meta-analysis using more refined genotype-based techniques, have confirmed this association and gone on to reveal a hierarchy of susceptibility and resistance alleles that map to the HLA system [30, 31]. The strongest linkage of anti-GBM disease is with HLA-DRB1*1501, being found 3.5 times more in patients than in the control groups. By excluding the effect of DRB1*1501, subsequent analysis demonstrated an increased frequency of DRB1*03 and DRB1*04 and a decreased frequency of DRB1*01 and DRB1*07. Furthermore, an increased frequency was found of DQB1*06 (some alleles of which are in linkage disequilibrium with DRB1*1501) and DQB1*03 and negative associations with DQB1*05 alleles. Similar HLA associations have now been reported in Japanese populations [32].
The recent meta-analysis [31] demonstrated that both inherited DRB1 alleles exert an effect on disease susceptibility, but that if one of these alleles was DRB1*1501, the effect of the second allele was either neutral (DRB1*04 and DRB1*03) or diminished the susceptibility to disease (DRB1*01 and DRB1*07) caused by DRB1*1501. Moreover, by comparing the structure of the susceptibility (DRB1*1501) and protective (DRB1*0701) alleles, the authors found that a particular region of the peptide-binding groove of the HLA-DRB1 (pocket 4) was significantly different and may therefore play a role in disease susceptibility. Interestingly, protective DRB1*07 and DRB1*01 alleles bind the majority of peptides corresponding to α[alpha]3(IV)NC1 sequence with greater affinity than the DRB1*15 alleles [33]. This suggests that the mechanism of disease susceptibility with DRB1*15 is not due to more efficacious presentation of an α[alpha]3(IV)NC1-derived peptide but may be due to competition for the autoreactive peptide epitope by the protective (DRB1*07 and DRB1*01) alleles. However, the precise molecular basis of disease susceptibility conferred by HLA-DRB1*15 alleles remains unknown.
Non-HLA Genes
Experimental anti-GBM models demonstrate that Th1 predominant strains appear to be susceptible to disease induction whereas Th2 strains are resistant, while other non-major histocompatibility (MHC) genes also confer susceptibility since disease can be induced in some, but not other, strains carrying the same MHC alleles [34]. These differences do not appear to be related to levels of expression of α[alpha]3(IV)NC1 in the thymus, nor to differences in the COL4a3 gene, which encodes rat α[alpha]3(IV)NC1 [35].
A role in maintaining B cell tolerance to the autoantigen has recently been shown for the inhibitory FC gamma receptor IIB (FCλ[gamma]RIIB), since mice deficient in this receptor developed severe pulmonary and renal lesions following immunization with type IV collagen, unlike wild-type animals [36]. In addition, recent data from patients with anti-GBM disease demonstrates that FCλ[gamma]RIIB polymorphisms(232T/I) are present at significantly elevated frequencies when compared with healthy controls [37], at least in Chinese populations. While the inhibitory receptor appears to attenuate disease, deficiency of all FC λ[gamma] receptors (FCRλ[gamma]) is associated with diminished disease, suggesting that stimulatory receptors are necessary for disease induction [38] and indeed copy number variation of the activatory FcgRIIIA receptor is found at higher frequency in patients compared to matched controls [39].
Environmental Factors
Although there is a strong disease association with HLA-DRB1*1501, this allele is present in almost a third of the white population and thus cannot solely explain the susceptibility to such a rare disease. It has therefore been proposed that a “second hit” occurs in susceptible individuals, which may be genetic or environmental.
In his original description of the clinical condition, Goodpasture believed he was studying the pathology of influenza infection, and although it has been shown that infectious episodes may precede clinical signs of autoimmunity [40], identifying a specific etiological infectious agent has not been successful.
Pulmonary hemorrhage in Goodpasture’s disease has been associated with a history of cigarette smoking, and disease relapses have been temporally linked with episodes of smoking [41]. Furthermore, there are human and experimental data suggesting that disease may be related to inhalation of hydrocarbons, although in some cases the exposure may simply have provoked a manifestation of an already existing disease [42, 43]. It is possible that pulmonary injury (through cigarette smoke or inhaled hydrocarbons) exposes the cryptic epitope within the alveolar basement membrane, thus triggering an immune response towards the α[alpha]3(IV)NC1 with subsequent anti-GBM disease. This suggestion is indirectly supported by the finding that exposure of α[alpha]3(IV)NC1 hexamers to reactive oxygen species is followed by significant anti-GBM antibody binding, which is absent using intact hexamers [44].
Autoimmunity
Humoral Immunity
The first demonstration that anti-GBM autoantibodies may be pathogenic dates back to 1967 when, in a classic series of experiments, Lerner, Glassock, and Dixon transferred antibodies eluted from the kidneys of patients with Goodpasture’s disease to squirrel monkeys, which subsequently developed proliferative glomerulonephritis [45]. Adoptive transfer experiments such as these have been repeated using a number of animal models demonstrating that serum from diseased animals to syngeneic recipients leads to development of disease [46]. The pathogenicity of the autoantibodies is supported by clinical data from patient studies. For example, rapid disease recurrence occurs if transplantation is performed in the presence of circulating anti-GBM antibody [47], and there are correlations between disease severity and antibody titer [48].
The majority of patients’ anti-GBM antibodies bind to the non-collagenous-1 domain of the α[alpha]3 chain, and in some cases the α[alpha]5 chain, of type IV collagen (α[alpha]3(IV)NC1 and α[alpha]5(IV)NC1, respectively), molecules that are found only in specialized basement membranes in sites such as the kidney, lung, choroid plexus, retina, and cochlea [47, 49, 50]. The clinical features are therefore a result of the tissue distribution of the antigen with pulmonary and renal disease predominating [51]. Rare cases with neurological involvement have been reported, but it is not clear if these are due immune effects on cerebral targets (such as the choroid plexus) [52]. The α[alpha]3 chain forms a triple helix with the α[alpha]4 and α[alpha]5 chains, which combines with another triple helix, through the NC1 domains, to form a NC1 hexamer, reinforced through disulphide bonds. Some patients may also form additional antibodies directed towards other collagen chains (such as the α[alpha]1, α[alpha]4, or α[alpha]6 chains of type IV collagen) [49, 53, 54]. The majority of the autoantibodies are of IgG1 subtype [55], while data from Chinese patients suggest that greater degrees of renal impairment are associated with a greater proportion of IgG1 and IgG3 [56]. The specificity of circulating and tissue-bound antibodies is identical, shown following elution of antibodies from affected organs [54]. Two immunodominant antigenic epitopes have been identified, termed epitopes A(EA) and B(EB) [57–59] in α[alpha]3(IV)NC1, to which the antibody binds. These epitopes are in close proximity in the intact molecule and contain a high degree of homology, leading to predictions of high degrees of autoantibody cross-reactivity and stabilization occurring, which may explain the high-affinity interaction [60]. Although antibodies may bind these sites separately, some autoantibodies recognize an epitope formed by combination of both antigenic sites [61]. Importantly, anti-GBM antibodies do not bind the intact NC1 hexamer but do so only following its dissociation [61]. Moreover, it appears that in vivo the epitopes are sequestered under normal circumstances, by α[alpha]4 and 5 chains, and only exposed following some physicochemical disruption of the GBM—such as exposure to hydrocarbons, smoking, infection, lithotripsy [62], degradation by reactive oxygen species [44], or basement membrane disruption consequent to some other glomerulonephritis [11, 63]. These may disrupt the NC1 hexamer and in turn induce conformational changes in the EA and EB epitopes, allowing priming of the immune response [54]. This is in contrast to anti-GBM disease that can arise in transplanted kidneys in patients with Alport’s disease, in whom the α[alpha]5 molecule is recognized as foreign and in which the alloantibodies can bind the epitope within the intact hexameric structure [54].
Cellular Immunity
Autoantibodies may be sufficient to transfer disease to primates, but in other model systems, cellular-based immunity is necessary, since transfer of autoantibodies to T cell receptor knockout mice does not result in disease, and disease can be transferred using mononuclear cells in both murine and avian models [46, 64]. Furthermore, generation of anti-GBM antibodies that bind in the kidney occurs in both susceptible and resistant strains of mice following immunization [65]. Thus, although autoantibodies are centrally important to disease pathogenesis, autoreactive T cells appear to be both necessary and sufficient to induce disease in animal models.
Evidence demonstrating a role for cellular immunity in human disease pathogenesis include the striking HLA-DRB1*1501 and DRB1*04 association [30], the class-switched (IgG1 and IgG4) autoantibody isotypes in the majority of patients (implying T cell-mediated help), and the finding of both CD4+ and CD8+ T cells within affected glomeruli [66, 67]. Furthermore, in rodent models, disease can not only be transferred by mononuclear cells but is also inhibited by anti-T cell therapies, including CTLA4-Ig co-stimulatory blockade [68] anti-CD4 and anti-CD8 monoclonal antibodies, and cyclosporin A [69]. Disease is absent in CD4+ and CD8+ knockout animals, requires a Th1 pattern of cytokine expression [65], and is attenuated following oral administration of GBM, inducing a form of mucosal tolerance [70]. Moreover, it appears that disease can be dissociated from autoantibody generation, since a number of animals may develop anti-GBM autoantibodies but neither nephritis nor alveolitis, while disease in others may be accompanied by circulating but not deposited anti-GBM antibody [71].
Evidence of autoreactive T lymphocytes directed against the α[alpha]3(IV)NC1 antigen has been demonstrated in patients by proliferation of peripheral blood mononuclear cells taken at the time of disease presentation [72]. Of note, healthy control subjects also demonstrated some proliferative capacity to these antigens, despite an absence of anti-GBM autoantibodies. This has been confirmed recently using peptides derived from α[alpha]3(IV)NC1, to which most control subjects had some response [73]. These data are similar to those described in multiple sclerosis patients and healthy subjects tested for T cell reactivity to myelin basic protein. However, there is a significantly different frequency of α[alpha]3(IV)NC1-specific autoreactive T cells in acute patients and controls [74]. Furthermore, this frequency diminishes with time from disease onset and reaches control levels after a number of years, mirroring the decline of anti-GBM autoantibodies in untreated patients.
The rarity of α[alpha]3(IV)NC1-specific autoreactive T cells in healthy individuals is in part due to the deletion of these cells during thymic ontogeny. In keeping with data on other tissue-specific autoantigens, a number of groups have found that human thymus expresses the α[alpha]3(IV)NC1 autoantigen, making it likely that negative selection of α[alpha]3(IV)NC1-reactive T cells does occur [74, 75]. Whether there is a difference in levels of α[alpha]3(IV)NC1 expression in the thymus between patients and healthy individuals, as has been suggested for other autoantigens (e.g. in human diabetes), has not yet been established.
The epitopes within α[alpha]3(IV)NC1 to which the autoreactive T cells respond to have been harder to define. From animal models a single peptide (containing three critical amino acids) is sufficient to induce disease, and this resembles peptides derived from a number of infectious agents, implicating molecular mimicry as a potential pathogenic mechanism and thus linking infection with subsequent disease [76]. Interestingly, the peptides that bind the susceptible HLA molecules most avidly are not the ones to which the T cells respond [77, 78]. Indeed rapid destruction of the autoreactive peptides by proteases (such as Cathepsin D) during antigen processing may serve to limit the exposure of autoreactive T cells to its antigen and may explain why these low frequency cells escape thymic selection [73].
Although the factors responsible for initiating the proliferation of autoreactive T cells in patients but not in healthy people remain unknown, they may relate to the exposure of neo-epitopes following NC1 hexamer dissociation, altered antigen processing allowing the pathogenic epitopes to be presented, or altered T or B cell regulatory mechanisms. This is in the context of the individual’s MHC class II alleles, as well as to other genetic and environmental factors.
Induction of Autoimmunity
The isolation of anti-GBM antibodies, albeit at low titer, and of circulating α[alpha]3(IV) NC1-specific T cells in healthy controls suggested that there may be a failure to appropriately delete these autoreactive cells during immunological development and that the cryptic nature of the autoantigen or the rapid degradation of the autoreactive epitopes prevents disease induction under normal circumstances. However, recent experiments using transgenic mice expressing α[alpha]3(IV) NC1-reactive immunoglobulin have demonstrated that autoreactive B cells are effectively deleted during maturation. These data suggest that generation of autoreactive B cells may result as a failure of central tolerance through IgG editing or following somatic mutation of mature B cells [79]. These data assumed that some autoantigen is expressed or delivered to the bone marrow during ontogeny. However, even in the absence of autoantigen (achieved by cross breeding the transgenic α[alpha]3(IV) NC1-Ig reactive mice with α[alpha]3(IV)-deficient animals), it appears that autoreactive B cells are eliminated during development, implicating a more complex regulatory network and suggesting that another (bone marrow-expressed) antigen may promote deletion of the α[alpha]3(IV) NC1-autoreactive B cells [80]. The nature of this potential tolerizing antigen remains to be elucidated. Interestingly, in patients (in whom tolerance has failed), this antigen or a cross-reactive antigen may be responsible for episodes of immune activation and disease.
Using a prospective analysis of sera collected from patients who had samples stored in the defense department repository, some of whom went on to develop anti-GBM disease, Olson et al. reported that asymptomatic anti-GBM antibodies are found in patients up to 10 months before diagnosis [5], but at low levels, up to many years in advance. They found a sudden increase in titer immediately preceding clinical disease. Interestingly, they also described low-level ANCA that preceded the development of anti-GBM antibodies in a significant proportion of patients, up to 5 years in advance, and postulated a possible pathogenic role for ANCA inducing GBM damage and exposing a cryptic epitope.
The need to break multiple regulatory mechanisms and expose the cryptic epitope may explain the rarity of clinical disease and the reason for a paucity of disease relapses, unlike other autoimmune diseases. Recent reports of anti-GBM disease arising following depletional biologic therapy with alemtuzumab (Campath-1H) also suggest that autoreactive B cells may be found in some susceptible individuals, but only when regulatory circuits are damaged do increased B cell responses arise followed by clinical disease [81].
Therapeutic Advances
Untreated, the disease was rapidly fatal in cases with pulmonary hemorrhage or in those with advanced renal failure prior to the availability of dialysis. However, with the introduction of dialysis, mortality rates were significantly reduced, and these were further improved, as was renal recovery, with the introduction of immunosuppressive regimens. In those patients presenting dialysis dependent and with 100 % crescentic change on renal biopsy, many practitioners avoid immunosuppression as renal recovery is unlikely, and the risks of immunosuppression outweigh the benefits [82]. However, in the face of other degrees of renal involvement, there is still a chance of regaining independent renal function [4] and this tips the balance in favor of treatment, assuming the patient is assessed as being robust enough to withstand immunotherapy. The other situation in which treatment may be appropriate in someone with dialysis dependency is if there is a living donor willing to donate a kidney, in which case more rapid antibody removal will be achieved (generally within 2–8 weeks) [8], allowing earlier transplantation (see below). Without immunosuppression, anti-GBM antibodies may persist for variable periods of time ranging from a few months to years [82].
Therapy with cyclophosphamide, corticosteroids, and plasmapheresis has remained the gold standard for over 30 years. Standard regimens utilize oral daily cyclophosphamide for 3 months; oral corticosteroids, tapered over 6 months; and plasmapheresis (Table 9.2). Whether pulsed intravenous cyclophosphamide is equivalent to daily oral therapy has not been demonstrated in any study, but as with dialysis-dependent ANCA-associated vasculitis, it appears some practitioners have already changed to this modality with various cases describing its use [52, 83, 84]. It would clearly be of interest to compare outcomes of oral and pulsed intravenous regimens, the latter of which clearly provides a benefit with regard to infectious risks and reduced cumulative doses of cyclophosphamide.
Table 9.2
Recommended treatment regimen for anti-GBM disease
Treatment | Dosing |
|---|---|
Plasmapheresis | • Daily 3–4 L exchanges for 5 % human albumin solution. Addition of fresh frozen plasma for patients undergoing invasive procedure (e.g., biopsy) within 72 h • Should be continued until antibody level is undetectable • Withhold if platelet count <70, fibrinogen <1 g/L • Watch for hypocalcemia, hypokalemia, and coagulopathy |
Prednisolone | Daily oral dosing at 1 mg/kg, with maximum at 60 mg. Reduce dose weekly to 20 mg by week 6, then more slowly. Aim to stop all steroids by 6 months |
Cyclophosphamide | Daily oral dosing 2–3 mg/kg. Reduce dose in older patients (1 mg/kg in >70 years). Withhold if WCC < 4 × 109 Pulsed intravenous cyclophosphamide may be as good but no evidence for this |
Prophylactic therapies | • Oral antifungals (nystatin/amphotericin or fluconazole)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




