Fig. 15.1
Thrombus formation in the kidney of aHUS. (a) Silver-methenamine stained section of a glomerulus of an aHUS patient with microthrombi (white arrow). Black arrow points at RBCs (×40). (b) Thrombus at vascular pole. Light microscopy: trichrome stain showing a thrombus at the vascular pole (white arrow). Note fragmented red cells in capillary lumen at tubular pole (black arrow) (trichrome ×40). (c) Intracapillary thrombus. Electron microscopy: fibrinoid material within glomerular capillary completely occluding the capillary lumen (×4700). Photos kindly provided by Sanjeev Sethi, MD, PhD, Mayo Clinic, Rochester, USA
TMA presents as a spectrum of related disorders with hemolytic anemia and thrombocytopenia. Multiple factors have been described which are related and may even sometimes cause identical pathological effects leading to thrombus formation. Thus, the two subtypes cause prominent damage of the endothelial lining and exposure of the subendothelial matrix that induces thrombus formation (Fig. 15.1b, c). The molecular causes for these defects are often linked to the defective regulation or inappropriate activation of the complement—as well as of the coagulation cascade. Both disease subtypes may have distinct initial triggers, and the affected modified molecules ultimately cause thrombus formation and endothelial damage. Despite similar presentations, the identification of the disease etiology, i.e., genetic or autoimmune, is important in order to tailor appropriate therapy. For example, the EHEC–HUS outbreak in Germany 2011 showed that deep knowledge of disease mechanisms although highly relevant may not apply to each individual case. In this chapter, we present recent scientific developments in the field of TMA and summarize the emerging common features of these complex disorders. TMA is an important example of translational medicine. Novel genetic causes have been linked to new pathological concepts and successfully transferred into clinical practice.
Thrombotic Thrombocytopenic Purpura
TTP is a severe life-threatening disease that exhibits symptoms like fever, microangiopathic hemolytic anemia, thrombocytopenia, and renal and neurological involvement and mainly affects women between 10 and 40 years of life [9–11]. In TTP, microthrombi rich in von Willebrand factor (vWF) develop in the arterioles and capillaries of the brain as well as other organs [9]. Both genetic and acquired autoimmune forms of TTP including hereditary mutations of the ADAMTS13 gene and autoantibodies that bind to ADAMTS 13 have been described [11, 12]. Both forms lead to platelet aggregation and microvascular thrombosis. A direct link between HUS and TTP is provided with the Escherichia coli Shiga toxin Stx1B and Stx2B, which induce cell signaling of human umbilical vein endothelial cells and secretion of von Willebrand factor [13].
The genetic form of TTP is associated with mutations in the coding region of the plasma metalloprotease ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type 1 repeats) gene [12, 14] (Fig.15.2a). The gene is primarily expressed in the liver, and the encoded plasma protease ADAMTS13 is a multidomain protein, which forms complex interactions with its substrates, cleaves large multimers of vWF, and thereby affects vWF function. The large vWF multimers cause platelet adhesion, platelet aggregation, and tissue infarction [12]. Endothelial and renal tubular cells also express ADAMTS13, and at the cell surface, the secreted protein is responsible for maintaining the cellular surface free of adhesive vWF [15, 16].
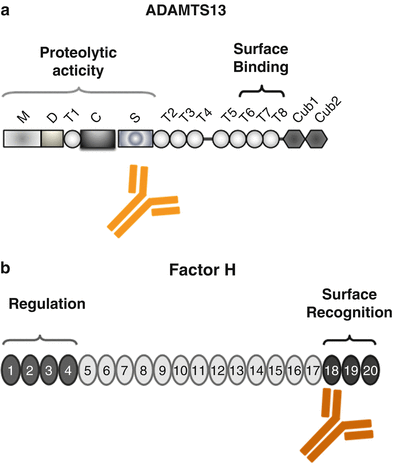
Fig. 15.2
Domain structure of ADAMTS13 and Factor H. (a) ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type 1 repeats) has a modular composition being formed of M: metalloprotease domains, D: disintegrin-like domain, T1: thrombospondin type 1 repeat, C: Cys-rich region, S: spacer region, and CUB-like domains 1 and 2. The position of the two major functional regions that form the proteolytic and surface-binding regions is indicated. The binding sites for the autoantibody are also shown. (b) Factor H is composed of 20 modules, termed short consensus repeats (SCRs). The central functional domains are located in the N terminus (SCR domain 1–4) forming the regulatory domain that controls complement activation at the level of the alternative pathway convertase C3bBb and in the C terminus (SCRs 18–20) that forms the surface binding and attachment region of the protein. The majority of aHUS-associated mutations in the Factor H gene are positioned in the C-terminal surface and attachment region and are mostly heterozygous. In DEAP–HUS which represents the autoimmune form (deficient for CFHR1 and CFHR3 proteins and autoantibody-positive form of HUS), autoantibodies to Factor H develop which bind to the C terminus and block C-terminal attachment to cell surfaces and biomembranes
Single-molecule experiments show that elongation forces in the vasculature unfold the A2 domain of vWF and that ADAMTS13 cleaves specifically the unfolded accessible A2 domain of vWF [17]. The structure of ADAMTS 13 and vWF provides important insights into the domain organization and allows us to map and define the interface of the two proteins. ADAMTS13 cleaves the Tyr1605–Met1606 bond in the central A2 domain of vWF. However, leukocyte proteases like elastin and protease 3 also cleave vWF close to ADAMTS13 cleavage site [11, 18]. The contact between ADAMTS13 and vWF is quite complex and several sites of interaction occur. The terminal linked sialic residues enhance proteolysis by ADAMTS13 [19, 20]. Both the thrombospondin1 Cys-rich region and the spacer domain of ADAMTS13 bind to the A2 domain of vWF [21]. A second ADAMTS13 binding site spanning residues 1874–2813 is located in the constitutively exposed D4 region of vWF [22]. In addition N-glycans are further relevant for proteolytic cleavage and for secretion [23].
ADAMTS13 gene mutations are spread over the complete gene and include several protein domains. Mutations, which result in intravascular platelet aggregation and thrombocytopenia, may result in defective substrate processing and deregulated stability of ADAMTS13 [24]. To illustrate the complexity, factor H gene mutations have linked TTP and complement. In two sisters from one family with similar mutations in the Factor H gene, one girl developed TTP but the other HUS [25].
The autoimmune form of TTP is characterized by the presence of autoantibodies that bind to ADAMTS13 and thereby block proteolytic degradation of the ultra large forms of vWF [26, 27]. The majority of autoantibodies bind within the spacer domain of ADAMTS13 (Fig. 15.2a). In addition the various IgG and IgM antibodies apparently influence the stability of the protease or inhibit ADAMTS13 binding to the surface of endothelial cells [28, 29]. IgG4 is the most frequent immunoglobulin subclass and is identified in about 90 % of the autoimmune-positive patients. The other subclasses include IgG1 (52 %), IgG2 (50 %), and also IgG3 (33 %). IgG4 autoantibodies may also occur in combination with other immunoglobulin subclasses. Whether and how the IgG subclasses are useful to predict disease recurrence requires further research [30].
Therapy: Genetic or acquired ADAMTS13 deficiency can be treated by plasma infusion or exchange. In addition hematopoietic progenitor cell-mediated gene therapy in ADAMTS13-deficient knockout mice was evaluated as novel therapeutic concept. This new model may form a basis for new therapy to treat genetic forms of human TTP [31].
Hemolytic Uremic Syndrome
HUS manifests primarily in the kidney and leads to acute renal failure together with microangiopathic hemolytic anemia and thrombocytopenia. At present three major forms of HUS are recognized: the classical also termed typical HUS, aHUS, the atypical form, and the autoimmune form DEAP–HUS [4, 5]. Thrombi are particularly formed in the kidney and are often associated with fragmented red blood cells. The concept of common pathogenetic mechanisms will be discussed as overlapping features, and combined factors are becoming evident between the disease subforms.
Typical HUS, also termed (D + HUS), is associated with diarrhea and is the most frequent form. This form occurs in approximately 80 % of HUS patients and is frequent in children. D + HUS is often caused by infections with EHEC, Shiga toxin-producing E. coli, Shigella dysenteriae type 1, and also with S. pneumoniae. Evidence is emerging that D + HUS can also be triggered by other infectious agents including a wide range of enterohemorrhagic E. coli, Bordetella pertussis, and viral infections like H1N1 influenza and Varicella zoster [32–34].
A role for complement in the pathogenesis of EHEC-induced typical HUS is demonstrated by the fact that Shiga toxin binds the central soluble complement inhibitor Factor H modulating the alternative complement cascade at local sites [35]. Similarly a role for the alternative pathway complement in typical HUS/D + HUS is demonstrated from a cohort study with 17 children showing increased levels of the complement activation product Bb and of the terminal complex SC5b-9 in patient’s plasma [36].
Organs affected and damaged by HUS are not limited to the kidney and include neurological, gastrointestinal, cardiac, and pulmonary manifestations in patients with typical, diarrhea-associated HUS. For patients with neurological symptoms, seizures, protracted severe seizures, as well as coma have been reported [6].
The EHEC outbreak in Germany during May to July 2011 was caused by enterohemorrhagic E. coli of the serotype O104:H4 that was distributed via contaminated sprouts. The incubation period for this E. coli strain was about 8 days and thus longer than the period of 4 days for the strain E. coli O157:H7. The infection rates were different between local states in Germany with the northern part of the country more affected. For some states the incidence for HUS was as high as 100 cases per one million populations. The majority of the infected individuals were adult females and neurological symptoms were frequently reported. A total of 2,987 infected individuals developed gastroenteritis and 18 patients (0.6 %) of this group died. In addition, 35 (4.1 %) patients of the 855 patients who developed HUS died (Robert Koch Institute report [37–40]). As typical HUS is caused by infections, the role of host predisposing genetic factors as well as determinants of infection susceptibility is an area for future research.
The second form of HUS, atypical (aHUS), also called non-diarrhea form of HUS (D-HUS), represents about 10–15 % of patients. The disease is more frequent in adults, but children are also affected. In addition to renal involvement, other organ and tissue involvements have been reported including cardiovascular ischemic events, stenosis of cranial arteries, as well as pulmonary and coronary arteries in patient with aHUS [4, 5].
aHUS is due to mutations in genes that code for complement regulators or complement components that affect the activity of C3bBb convertase of the alternative complement pathway. Most mutations in atypical HUS show incomplete penetrance, are heterozygous in nature, and affect one allele. Consequently patients have a second intact allele [41].
Regulators modulated in aHUS include the central fluid phase complement inhibitor Factor H (20–30 %) (Fig. 15.2b), the membrane-bound regulator MCP/CD46 (5–15 %), the soluble serine protease Factor I (4–10 %), and the two major components that form the alternative pathway convertase C3bBb, i.e., C3 (2–10 %) and Factor B (1–4 %). In addition, thrombomodulin, which is a regulator of the coagulation cascade and a likely effector of complement action and of the C3 convertase, has been also linked to aHUS [42, 43]. Which complement gene shows the mutation is an important parameter for the prediction of disease outcome, as well as for posttransplant recurrence.
Based on the strong involvement of complement gene mutations in more than 60 % of aHUS patients, this type of HUS is sometimes referred to as complement-HUS [41, 42, 44, 45]. In general terms heterozygous mutations in complement genes result in defective control of the alternative pathway convertase C3bBb and in widespread activation of the complement cascade. The defective regulation is thought to occur at specific sites, e.g., at the surface of renal endothelial cells, exposed subendothelial matrix, or on platelets. In addition a role for the terminal pathway of complement in aHUS is emerging. CFHR1, the gene that is deleted in DEAP–HUS, is a terminal pathway regulator that blocks the C5 convertase as well as assembly and membrane insertion of the terminal complement complex (TCC) [45]. Similarly, a mutation in the gene coding for the terminal complement inhibitor clusterin (A1298C—Q433P) has been identified in a child with typical HUS [46].
The third group of HUS, the autoimmune form DEAP–HUS (deficient of CFHR1 and CFHR3 proteins and autoantibody-positive HUS) [47, 48], is characterized by the presence of autoantibodies. Autoantibodies to Factor H are detected in about 10–15 % of all HUS patients, particularly in children age 4–17 years [47]. Autoantibodies to Factor H are of the IgG1 and IgG3 subtype [49]. In most cases these autoantibodies develop on a specific genetic background of homozygous deletion of a 84 kb chromosomal segment on human chromosome 1q32, which encodes the two genes CFHR1 (complement Factor H-related protein 1) and CFHR3. Consequently the two complement proteins CFHR1 and CFHR3 are absent in the plasma of these patients [8, 48]. Most IgG autoantibodies of DEAP–HUS patients bind to the C-terminal surface attachment region of Factor H, i.e., SCRs 18–20, and block Factor H binding to heparin and glycosaminoglycans and also inhibit Factor H recruitment to the surface of endothelial cells. The result is reduction in the local regulatory and anti-inflammatory functions of Factor H at modified cellular surfaces. Autoantibody-positive plasma derived from aHUS patients causes enhanced lysis of sheep erythrocytes, thus demonstrating directly that autoantibodies mediate complement regulation at cellular surfaces by influencing surface binding of Factor H [50, 51] (Fig. 15.2b). Importantly, the C-terminally binding autoantibodies and mAbs do not block or influence the complement regulatory activity of Factor H in fluid phase in plasma, which is mediated by the N-terminal region.
The frequency of autoantibodies and the correlation of autoantibodies with CFHR1–CFHR3 deficiency have been shown in the Jena-HUS, the Paris, the Newcastle, and in the Spanish HUS cohorts [52, 53]. In all four cohorts, the frequency for DEAP–HUS was between 6 and 14 %, and more than 90 % of the DEAP–HUS patients had a combination of autoantibodies together with homozygous chromosomal CFHR1–CFHR3 or CFHR1.CFHR4 deletion. In addition, new genomic rearrangements in the CFHR gene cluster were reported in various DEAP–HUS patients. Also in aHUS patients, chromosomal deletions that results in hybrid genes, e.g., a hybrid Factor H-CFHR1 gene as well as a hybrid Factor H-CFHR3 genes and hybrid Factor H-CFHR proteins have also been reported [54, 55]. Human chromosome 1 region 1q32, which includes several long repetitive regions, is frequently involved in recombination processes, resulting in novel chromosomal rearrangements. Furthermore genetic deletion variants have been identified in the genome of both aHUS and DEAP–HUS patients (Fig. 15.3) [48, 53]. Novel breakpoints such as the deletion of the CFHR1 gene combined with the presence of CFHR3 were described, and in a large pedigree with three aHUS patients, a hybrid Factor H-CFHR3 protein was identified (Fig. 15.3). This hybrid protein consists of 19 domains of Factor H (i.e., SCRs 1–19) and five SCR domains of CFHR3 (i.e., SCRs 1–5) and shows severely reduced complement regulation and increased heparin binding. The genetic cause was microhomology-mediated end joining [55].
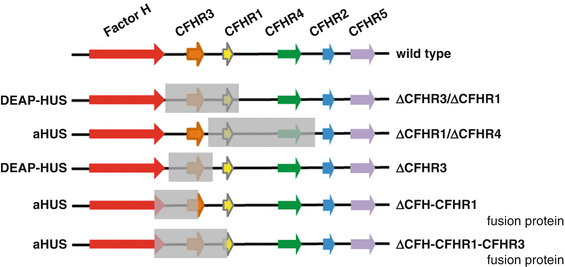
Fig. 15.3
Deletion in the Factor H gene cluster in aHUS and DEAP–HUS. The Factor H gene cluster is located on human chromosome 1 region 1q32 and includes the genes Factor H, CFHR3, CFHR1, CFHR4, CFHR2, CFHR5, and Factor XIIIb. The wild-type organization is shown in the top panel (wild type). The first rearrangement that forms a fusion gene between Factor H and CFHR1 is shown in the second line (conversion type). Homozygous deletion of a 84 kbp chromosomal fragment that includes the gene coding for CFHR1 and CFHR3 is frequently associated with the autoimmune form DEAP–HUS and in HUS correlates with the presence of autoantibodies to Factor H. However, this deletion is also observed in healthy persons, but is then not associated with autoantibodies. A new deletion was reported which leaves the CFHR3 locus intact and which results in deletion of the CFHR1 and CFHR4 genes (bottom line) [36]
CFHR1 and CFHR3 are both complement regulators; however, their mode of action and level of control are different from each other and also distinct regarding the regulation of Factor H. CFHR1 is a complement regulator that inhibits the C5 convertase, as well as assembly and membrane insertion of the terminal complement complex. CFHR3 is a complement regulator that acts as cofactor for factor I and allows degradation of C3b. Both CFHR1 and CFHR3 share similar and overlapping binding sites on the C3b protein with Factor H. As a consequence both CFHR proteins compete with Factor H for C3b binding as well as for attachment to human cell surfaces. By competing with Factor H ligand binding, each CFHR protein modulates the local activity of Factor H (Fig. 15.3). The coordinated binding suggests that at a cellular surface CFHR1 and Factor H (and likely CFHR3 and Factor H also) act in concert and control complement convertase and thus local amplification of the complement cascade in a sequential fashion [48, 54].
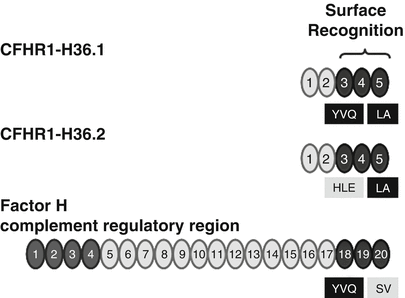
A polymorphism of the CFHR1 gene that was reported with the original cloning of the CFHR1 cDNAs is also associated with HUS. Two major CFHR1 variants differ by three amino acids in SCR 3. The CFHR1 variant encoded by cDNA H36.1 represents CFHR1-HLE and clone H36.2 encodes the CFHR1-YVQ variant. These isoforms appear to have different electrophoretic mobilities. The basic variant with residues H157, L159, and E175 (CFHR1-HLE) corresponds to the sequence of SCR 18 of Factor H. The sequence of the acidic CFHR1-YVQ variant differs from the sequence of Factor H (Fig. 15.4). The mostly C-terminal SCR domain, i.e., SCR 5, is identical for the two CFHR1 variants. However, between CFHR1 and Factor H this most C-terminal SCR differs by two residues. CFHR1–SCR 5 uses L290 and A296 and Factor H-SCR 20—the corresponding residues are S1191 and V1197 [53] (Fig. 15.4). Thus, the C-terminal surface-binding region of the original CFHR1 cDNA (H36.1) encodes the CFHR1-YVQ variant [54] and differs by only two residues (i.e., L290 and A296) with the corresponding C-terminal surface binding of Factor H. The allele frequency of the acidic variant CFHR1-HLE is 0.57 in a Spanish AMD cohort and is higher as in the control group (0.39). It will be of interest to define whether these three residues in SCR 3 affect the function and ligand interaction of the C-terminal surface recognition region.