1. Nonimmune fetal hydrops
2. Antecedent history in a newborn infant of pregnancy complications such as HELLP 3. syndrome or fatty liver of pregnancy
3. Unexpected deterioration and death of a normally developed newborn baby
4. Failure to thrive in an infant with abnormal facies and/or brain and skeletal anomalies
5. Sudden infant death syndrome (crib death)
6. Unexplained metabolic crisis precipitated by a mundane viral infection with or without a history of a previous episode
7. Unexplained organomegaly/organ dysfunction
8. First-degree relative with unexplained serious illness, single organ dysfunction or sudden death
Metabolic crisis: features
1. Hypoglycemia, nonketotic or hypoketotic
2. Normoglycemic ketoacidosis
3. Metabolic acidosis, not otherwise specified
4. Hyper- or hyponatremia
5. Liver dysfunction with or without hyperammonemia
6. Encephalopathy (lethargy, stupor, or coma)
7. Vomiting
8. New onset seizure
9. Rhabdomyolysis
More common causes of acute metabolic crisis in infants
1. Beta oxidation defects, especially MCAD
2. Mitochondriopathies
3. Lipid transport defects, e.g., systemic carnitine deficiency and carnitine palmitoyltransferase deficiency
4. Glycogen storage diseases, especially types I and III
13.2 Handling a Liver Biopsy Specimen for Suspected Metabolic Disease
At least two cores of liver tissue should be obtained. Cautery of surgical specimens must be avoided, or enzyme studies may be useless. Freeze one core of liver tissue or one slice of a surgical biopsy specimen in anticipation of need for biochemical or genetic analysis. Delay the decision about use of frozen liver tissue until light and electron microscopy are completed unless clinical data clearly indicate a particular class of metabolic disease. The value of electron microscopy is severely limited, if a sample of liver tissue is not placed in a suitable fixative such as glutaraldehyde without delay; 2 mm of a typical needle core is sufficient and need not be processed beyond embedding in resin if deemed unnecessary. If fixation for electron microscopy was overlooked, flash frozen tissue thawed in glutaraldehyde is superior to paraffin-embedded tissue for study of ultrastructure. When clinical suspicion of a metabolic disease/storage disease is supported by light microscopic findings, the decision of how to proceed is aided by assessment of ultrastructure of all cell types and organelles and evaluation for storage material and its location. For example, when light microscopy identifies features of glycogen storage disorder, and study of liver ultrastructure confirms that excess nonlysosomal glycogen has displaced structurally normal organelles in most hepatocytes, it is then appropriate to commit the frozen sample to measurement of the glycogen concentration and to screen for glycolytic enzyme defects.
13.3 Analysis and Reporting of Liver Biopsy Specimens for Suspected Metabolic Disease
Conventional stains of paraffin sections of a diagnostic liver specimen are hematoxylin and eosin, Masson trichrome, reticulin, and periodic acid-Schiff (PAS) with prior diastase digestion. Stains that prove useful only in certain contexts, but are nonetheless often routinely employed, are PAS, Prussian blue for hemosiderin deposits, and rhodanine for copper complexes in secondary lysosomes. Stains that help identify a stored material are Sudan black and Ziehl-Neelsen for ceroid and Alcian blue and colloidal iron for mucopolysaccharide or polyglucosan. Specific immunostains may help identify a particular protein such as alpha-1-antitrypsin or fibrinogen or a specific organelle such as mitochondria or lysosomes.
Preparation of a useful written report of a liver biopsy is a skill that can be acquired through practice. An appreciation of normal liver histology combined with orderly enumeration of histological features, both normal and abnormal, is essential. When chronic hepatitis of any kind is present, activity and stage should be assessed. Interaction with the clinician who performed the procedure is required to understand why it was done and what information is desired. The objective, whenever possible, should be diagnostic interpretation rather than mere description.
13.4 Histological Patterns of Metabolic Liver Disease
13.4.1 Normal or Near Normal Liver Histology in Metabolic Disease
Rarely, metabolic diseases that affect the liver are pure functional disorders with minimal or absent histological changes (Table 13.2). In certain circumstances, such a liver may be utilized for transplantation (Popescu and Dima 2012). Examples of metabolic diseases due to abnormal liver metabolism that consistently lack morphological abnormalities are primary oxalosis, acute intermittent porphyria, maple syrup urine disease, and homozygous hypercholesterolemia. Also lacking progressive liver disease are disorders of bilirubin conjugation (Gilbert syndrome, Crigler-Najjar syndrome types I and II), in which diminished or absent glucuronosyltransferase activity results in indirect-reacting hyperbilirubinemia without hemolysis (Sampietro and Iolascon 1999). Bilirubin encephalopathy is a hazard in the severe form of this disease. In Dubin-Johnson syndrome, mutations in the MRP2 gene impair secretion of conjugated bilirubin across the canalicular membrane into bile causing mild conjugated hyperbilirubinemia without liver disease. A melanin-like lipofuscin pigment accumulates in the apical region of zones 2–3 hepatocytes and secondarily in Kupffer cells, without further histological or functional consequences (Fig. 13.1).
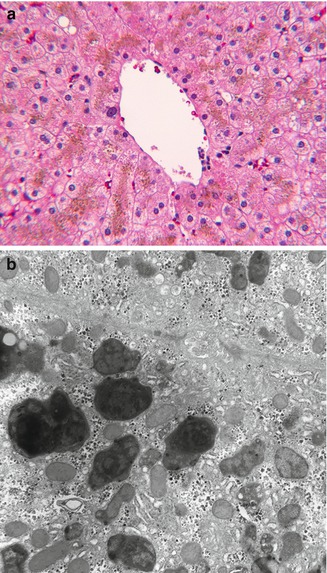
Table 13.2
Hepatocentric metabolic disease without chronic liver disease
1. Oxalosis |
2. Dubin-Johnson syndrome |
3. Crigler-Najjar syndrome |
4. Fabry disease |
5. Urea cycle defects |
6. Aminoacidemias |
7. Organic acidemias |
8. Disorders of glycosylation |
9. Alpha-1-antitrypsin phenotypes other than ZZ and SZ |
10. Smith-Lemli-Opitz syndrome |
11. Peroxisomal disease other than Zellweger disease |
Fig. 13.1
(a) Dubin-Johnson syndrome. Golden brown pigment accumulates in apical cytoplasm of zone 3 hepatocytes. H&E stain. (b) Bulky polymorphous residual bodies (secondary lysosomes) adjacent to canaliculus. EM
13.4.2 Liver Inflammation in Metabolic Disease
Idiopathic neonatal hepatitis (INH) is an umbrella term originally devised for transient nonobstructive cholestatic liver disease in the first several months of life. Histological features are lobular cholestasis, giant cell transformation of variable extent, persistent extramedullary hematopoiesis, mild inflammation, minimal or absent changes in bile ducts, and little or no fibrosis (Fig. 13.2). Modern advances in understanding of the etiology of cholestatic liver disease in infants with direct hyperbilirubinemia indicate the lesion of INH exists in numerous contexts and is often the initial histological diagnosis in an infant who develops progressive liver disease due to a specific metabolic disease (Table 13.3). Despite these advances, experience indicates that INH continues to have multiple etiologies including perinatal virus infection, immunopathy such as hemophagocytic lymphohistiocytosis (HLH), presumably undiscovered disorders of bile formation and transport, and as a transient expression of immaturity of the infant liver under stress. Histological features that suggest that an INH-like disorder may have a specific, underlying metabolic etiology are prevalence of pseudoacini, excessive giant cell necrosis, excessive inflammation, intralobular (pericellular) fibrosis, steatosis, prominent vacuolation of Kupffer cells, and prevalence of injury to small bile ducts. Liver ultrastructure in INH demonstrates nonspecific features of lobular cholestasis (Fig. 13.3) and may help identify specific metabolic diseases that cause direct hyperbilirubinemia in infants.
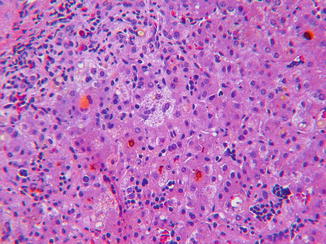
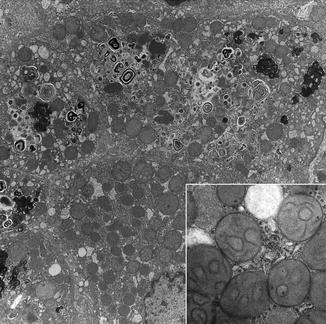
Fig. 13.2
INH. Canalicular and cytoplasmic cholestasis (cholate stasis), erythropoiesis in sinusoids, and myelopoiesis in portal area and focal giant cell transformation. H&E stain
Table 13.3
Conjugated hyperbilirubinemia in early infancy
1. Idiopathic neonatal hepatitis |
(a) Nonspecific stress/liver immaturity |
(b) Infection |
(c) Parenteral nutrition related |
(d) Endocrinopathy (thyroid, pituitary) |
(e) Unrecognized defect in bile formation or transport |
2. Biliary atresia |
3. Alpha-1-antitrypsin storage disease |
4. Alagille syndrome |
5. PFIC, especially PFIC 2 |
6. Galactosemia |
7. Fructose intolerance |
8. Mitochondriopathy |
9. Bile acid synthetic defects |
(a) 3-OH steroid dehydrogenase deficiency |
(b) 5-beta reductase deficiency |
10. Tyrosinemia |
11. Niemann-Pick disease, especially type C |
12. Zellweger syndrome |
13. Smith-Lemli-Opitz syndrome |
14. Cystic fibrosis |
Fig. 13.3
INH. Hepatocytes contain abundant secondary lysosomes and autophagic bodies. Membranous profiles suggest phospholipid derived from bile or autophagy. Inset: mitochondria show internal rearrangements of cristae. EM
Portal and lobular inflammation, other than extramedullary hematopoiesis, is usually a minor feature in INH and may be absent in many metabolic disorders that affect the liver of infants. The explanation may be lack of metabolic by-products that are toxic to the affected cells; or a toxic effect may be easily compensated by replacement of injured cells or organelles without permanent injury to the liver. On the other hand, some metabolic diseases of the liver are commonly accompanied by inflammatory changes, such as alpha-1-antitrypsin deficiency, tyrosinemia, and several bile acid synthetic defects, that may mimic chronic viral hepatitis or drug-induced hepatitis or autoimmune hepatitis. The common denominator in metabolic disorders consistently presenting as “hepatitis” seems to be accumulation of toxic metabolites that stress hepatocytes or small bile ducts. An alternate explanation is that inflammation incidental to intercurrent events such as viremia, sepsis, systemic inflammatory disease, or drug exposure may be poorly tolerated in metabolic disease. Examples of metabolic liver diseases with propensity to inflammation and/or cirrhosis and long-term liability to neoplasia are listed in Table 13.4.
Table 13.4
Metabolic diseases with chronic liver disease
Chronic inflammation a common feature |
1. Tyrosinemia |
2. Bile acid synthetic defects |
3. Alpha-1-antitrypsin storage disorder |
4. Wilson disease |
Cirrhosis a common outcome |
1. Galactosemia |
2. Fructose intolerance |
3. Alpha-1-antitrypsin storage disease |
4. Tyrosinemia |
5. Glycogen storage disease, type IV |
6. Wilson disease |
7. Bile acid synthetic defects |
Increased risk for hepatic neoplasms |
1. Tyrosinemia |
2. Glycogen storage diseases |
3. Alpha-1-antitrypsin storage disease |
4. PFIC2 |
13.4.3 Metabolic Disease with Prominent Lobular Cholestasis
Canalicular cholestasis and cytoplasmic chole-stasis regularly occur in INH and in certain metabolic diseases. Disruption of the canalicular network as a result of necrosis or extensive giant cell transformation may contribute to nonobstructive cholestasis. Examples are tyrosinemia, alpha-1-antitrypsin storage disease, perinatal iron storage disorder, galactosemia, and some mitochondriopathies. Another group includes disorders in which the synthesis or transport of bile acids is abnormal. In both, lobular cholestasis is severe, but in synthetic defects, toxic hydrophobic monohydroxy bile acids may injure bile canaliculi and ductules as well as hepatocytes. Bile ducts are spared and serum gamma glutamyl transferase is typically normal. A third group with cholestasis are genetic metabolic diseases that are characterized by progressive cholangiopathy affecting bile ducts such as Alagille syndrome, Zellweger disease, cystic fibrosis, and one form of progressive familial intrahepatic cholestasis, PFIC3. In this group are those few infants with alpha-1-antitrypsin storage disease who have early-onset cholestasis with progressive small bile duct injury potentially resulting in paucity. Serum gamma glutamyl transferase is typically elevated in cholangiopathies.
Paucity of interlobular bile ducts is by definition a feature of Alagille syndrome. Nonsyndromatic paucity has been reported rarely in a host of other metabolic, genetic, and acquired diseases such as alpha-1-antitrypsin deficiency, PFIC2, bile acid synthetic defects, congenital panhypopituitarism, or conditions such as Zellweger disease, Down syndrome, and arthrogryposis-renal-cholestasis syndrome. Paucity may be due to bile duct destruction with or without sclerosis or delays in small bile duct formation (Kahn et al. 1986; Sinha et al. 2007).
13.4.4 Bile Ductules Versus Ducts in Metabolic Disease
Recognition of the difference between proliferation of interlobular bile ducts and reactive bile ductules is important for distinguishing a true cholangiopathy such as biliary atresia from the common periportal ductular reaction (DR). The latter occurs in diverse liver diseases (viral, autoimmune, drug induced, and metabolic) that are not primary diseases of bile ducts (Fig. 13.4a–c). Persistent ductular reaction is a sign of injury to bile ductules and zone 1 hepatocytes. Metabolic diseases such as tyrosinemia, galactosemia, alpha-1-antitrypsin deficiency, and the more hepatotoxic bile acid synthetic defects are regularly accompanied by ductular reaction and progressive periportal fibrosis. Ductular reaction in Alagille syndrome and Zellweger syndrome may hinder recognition of paucity of interlobular bile ducts.
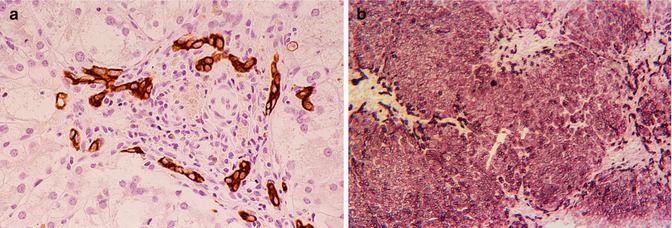
Fig. 13.4
(a) Ductular reaction. Interlobular bile duct inapparent though paucity was absent. 3-beta OH steroid dehydrogenase deficiency (bile acid defect). Cytokeratin immunostain. (b) Ductular reaction. Paucity of interlobular bile ducts is striking. Zellweger disease. Cytokeratin immunostain
True cholangiopathies are characterized by primary injury to intra- and/or extrahepatic bile ducts and are often accompanied by a persistent DR with progressive periportal fibrosis. DR and proliferation of interlobular bile ducts often coexist in large duct cholangiopathies, e.g., extrahepatic biliary atresia, ABCB4 disease (PFIC3), Langerhans cell histiocytosis, and primary sclerosing cholangitis.
Bile duct epithelium usually is not involved in metabolic storage diseases that affect Kupffer cells and/or hepatocytes. Exceptions include some lysosomal storage diseases (type II glycogenosis, GM1 gangliosidosis, mucolipidoses) and cystic fibrosis. In perinatal hemochromatosis (PH), hemosiderin deposits in duct epithelium may be observed possibly reflecting the general deposition of hemosiderin in extrahepatic parenchymal cells.
13.4.5 Acquired Steatosis Versus Metabolic Disease with Steatosis
Fatty change in the liver of infants and children is usually acquired but may be a manifestation of an underlying rare genetic metabolic disease such as a defect in beta oxidation of lipids, mtDNA depletion, disorder of specific electron transport proteins, urea cycle defect (UCD), organic acidemia, or an aminoacidopathy. Accumulation of lipid in hepatocytes without inflammation, hepatocyte necrosis or other abnormalities of liver cells, or evidence of fibrosis, is simple steatosis (Fig. 13.5a). Simple steatosis may be seen with cystic fibrosis, Wilson disease, celiac disease, protein avoidance in urea cycle defects, corticosteroid therapy, certain drug reactions, hypertriglyceridemia, after gastric bypass surgery and portocaval shunts, and with blind loop and short gut syndromes. Simple steatosis is characterized by large vacuoles of neutral lipid that displace the nucleus to the periphery (Fig. 13.5a). This change commonly is limited to periportal hepatocytes (zone 1) but may be panlobular, or limited to zone 3 in certain circumstances related to gastrointestinal dysfunction (Fig. 13.5b). A form of simple steatosis is readily acquired in infants and children during periods of temporary caloric deprivation. Simple histiocytosis is common at autopsy of critically ill children who die in hospital intensive care units and occurs in a minority of infants who are diagnosed with sudden infant death syndrome. In these circumstances, lipid droplets are of small to medium size, often panlobular, and tend not to displace hepatocyte nuclei (Fig. 13.5c). Because coalescence of lipid into visible medium- and large-sized vacuoles is a dynamic process, hepatocytes containing medium- and large-sized lipid droplets often coexist. Nonetheless, the predominant physical form of lipid vacuoles provides a clue, albeit an imperfect one, to the tempo and the underlying cause of lipid accumulation.
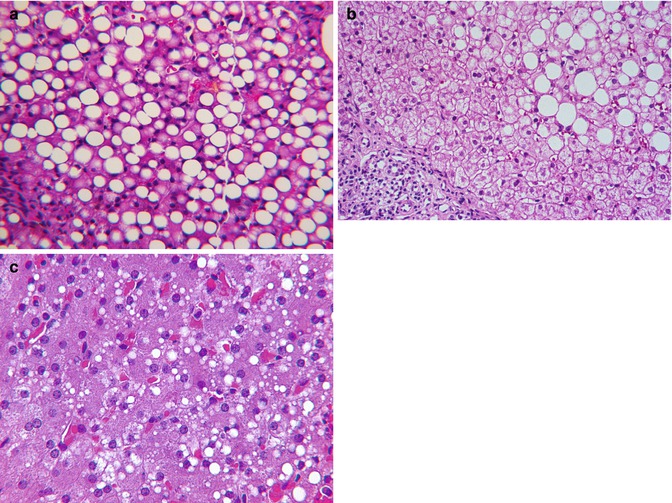
Fig. 13.5
(a) Simple steatosis. Cystic fibrosis with protein malabsorption. (b) Simple steatosis in GI tract disease. (c) Subacute simple steatosis at autopsy
Inflammation and fibrosis typically are absent in simple steatosis except in cystic fibrosis where focal obstructive cholangiopathy may coexist. Because lipid has been extracted during processing, it can only be presumptively identified in paraffin sections based upon the sharp interface with surrounding hepatocyte cytoplasm. Storage lysosomes may have a similar sharp interface by light microscopy, but ultrastructure discloses a limiting membrane.
Simple steatosis may be the only change in nonalcoholic fatty liver disease (NAFLD), or these patients may have steatohepatitis (SH) (Schwimmer et al. 2005). NAFLD is linked to obesity, type 2 diabetes mellitus, and insulin resistance. Obese children who develop transaminasemia are commonly subjected to liver biopsy for the purpose of staging and grading the liver lesion (Kleiner et al. 2005; Brunt et al. 2011). SH usually can be distinguished from simple steatosis or steatosis due to a genetic defect in lipid metabolism by subtle lobular inflammation and active hepatocyte injury. Findings in SH in addition to macro- and microsteatosis include multifocal lobular inflammation, ballooned degenerate hepatocytes, and, especially in children, portal inflammation (Fig. 13.6). Mallory-Denk bodies are more common in adults than children with SH. Ultrastructural changes in NAFLD are more commonly observed in SH than with simple steatosis. Excess smooth and rough endoplasmic reticulums are common but nonspecific. An array of focal mitochondrial changes presumably due to oxidative stress includes pleomorphism, matrix crystalloids, and occasional megamitochondria (Fig. 13.7). Persistent hepatocyte injury in NAFLD carries risk for progressive fibrosis but is reversible.
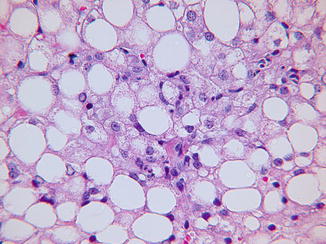
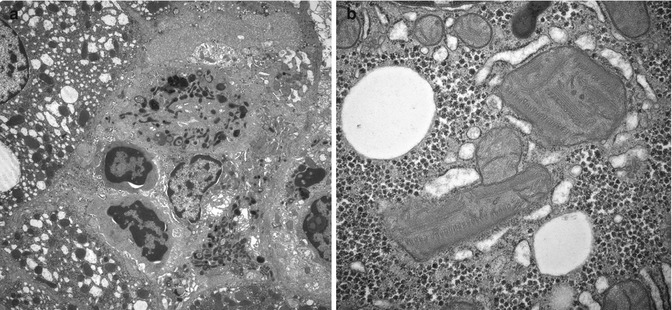
Fig. 13.6
Nonalcoholic steatohepatitis. Lobular inflammatory foci accompany fatty change. H&E stain
Fig. 13.7
(a) Nonalcoholic steatohepatitis. Sinusoidal inflammatory cell aggregates are common. Hepatocytes exhibit mitochondrial pleomorphism and universal dilatation of endoplasmic reticulum. EM. (b) Steatohepatitis. Stressed mitochondria contain paracrystalline matrix inclusions. EM
13.4.5.1 Steatosis, a Confounding Variable
Hepatic steatosis may coexist as a confounding variable in many metabolic disorders, either because of poor nutrition, or because an underlying genetic defect secondarily interferes with lipid processing or utilization (Table 13.5). TPN initiated before a specific diagnosis is made may contribute to steatosis. In MCAD and in some aminoacidopathies such as isovaleric acidemia, hepatocyte lipid accumulation may be prominent at the time of a metabolic crisis. Large vacuolar lipid accumulation in hepatocytes is common in type I and type III glycogenosis and rarely is so extensive as to obscure the diagnostic histological features of glycogen excess (Fig. 13.8).
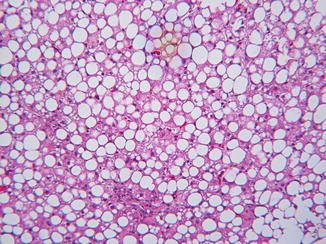
Table 13.5
Metabolic disease and steatosis
Steatosis a constant feature |
1. MCAD |
2. LCAD |
3. Galactosemia |
4. Fructose intolerance |
5. Citrin deficiency |
6. Glycogen storage disease, types I and III |
7. Abeta- and hypobetalipoproteinemia |
Steatosis an inconstant feature |
1. Cystic fibrosis |
2. Mitochondriopathy, especially congenital lactic acidosis |
3. Urea cycle defects |
4. Wilson disease |
5. Aminoacidopathies |
6. Glycogen storage diseases, other than types I and III |
Fig. 13.8
Steatosis, a common feature in GSD I, may mask glycogen excess. H&E stain
13.4.5.2 Steatosis in Neonatal Hepatitis Syndrome
Many metabolic liver diseases that present in infants are routinely included in the lengthy differential diagnosis of direct hyperbilirubinemia due to “neonatal hepatitis,” but it is helpful to know that with the following exceptions prominent lipid vacuolation of hepatocytes is unusual in most disorders now recognized as specific causes of neonatal cholestasis with giant cell transformation. Metabolic diseases presenting in young infants that often contain noteworthy amounts of lipid in hepatocytes include galactosemia; hereditary fructose intolerance; hepatic mitochondriopathies; urea cycle defects; Niemann-Pick A, B, and C; citrin deficiency; and lysinuric protein intolerance; in these disorders, lipid vacuoles of variable size may be accompanied by mild portal inflammation, progressive portal fibrosis, and liver failure. In all forms of Niemann-Pick disease, Kupffer cells are vacuolated as well as hepatocytes.
13.5 The Metabolic Disease Autopsy
When an infant or young child dies suddenly or after a brief hospitalization, without a specific diagnosis, and is suspected to have a metabolic disease, a common question is what investigations to pursue as part of the autopsy (Olpin and Evans 2004). Issues of cost and reimbursement eventually emerge, but the most essential component of any such investigation is availability of frozen tissue from heart, liver, and skeletal muscle and body fluid specimens such as urine, blood, bile, vitreous, and cerebrospinal fluid. Specimens collected during life and briefly retained in the clinical laboratory should be sequestered and frozen, along with those collected at autopsy. Additionally, a fibroblast culture should be established pending a decision about further investigation. Cultured fibroblasts contain many enzymes that are deficient in metabolic disease and are an invaluable source of DNA. The final decision about how to proceed is best guided by the clinical and family history and by the histological findings. Microscopy of organs critical to the particular questions posed such as heart, lungs, liver, and skeletal muscle should be expedited to guide the investigation. If an alternate explanation for illness/death is not found, and if histological findings suggest the presence of a metabolic disease affecting one or more organs such as the liver or heart, urine carnitine and acylcarnitine profiles and serum amino acid profiles may be helpful. The results of neonatal screening should be reviewed. Consider using an autopsy blood spot on a Guthrie card to obtain a repeat screen at a government or private laboratory that has a comprehensive newborn screening program.
The most likely causes of lethal metabolic crisis are listed in Table 13.3. The yield for the metabolic autopsy has not been high except for cases of congenital lactic acidosis and fatty acid oxidation defects; only the latter are associated with sudden unexpected death. However, new applications such as targeted or whole exome sequencing may change the outcome by alerting caregivers to risk, should they become economical.
13.6 Fatty Acid Oxidation Defects
13.6.1 Clinical Manifestations
More than a dozen disorders of in fatty acid oxidation (FAO) have been recognized (Vockley and Whiteman 2002; Saudubray et al. 1999). Morbidity/mortality is highest during infancy. A metabolic crisis with hepatomegaly and nonketotic hypoglycemia is a typical presentation in medium-chain acyl-coenzyme A (CoA) dehydrogenase deficiency (MCAD), the most common FAO, but true hepatic failure is rare. Progressive cardiomyopathy and episodic rhabdomyolysis are less common presentations of FAO.
MCAD is the most common of the defects in FAO. The incidence is about 1/10,000 live births in Caucasians but is much less common in Asians based upon data from newborn screening programs (Horvath et al. 2008). Newborns with high C8-acylcarnitine levels have an increased frequency of the common 985A > G mutation. Early detection by screening improves outcome because it highlights the vulnerability to fasting and need for preventative measures, particularly during the first several years of life. However, genetic variants of uncertain significance have emerged from the screening data, and genotype-phenotype correlation remains uncertain (Lindner et al. 2010). In MCAD, an acute metabolic crisis with nonketotic hypoglycemia is precipitated by a febrile illness that causes loss of appetite, exposing the inability of affected children to metabolize lipid during brief starvation. MCAD is also associated with cardiac arrhythmias and sudden death, accounting for a small percentage of neonatal and unexplained infant deaths. FAO disorders may mimic epidemic post-viral Reye syndrome (RS) in clinical presentation, although the affected age groups substantially differ. Morphological manifestations may provide a basis for differentiation if liver tissue is available for both light and electron microscopy (Treem et al. 1986).
Long-chain acyl-CoA dehydrogenase deficiency results in progressive liver fibrosis, often with cardiomyopathy, myopathy, or pigmentary retinopathy. FAO defects or lipid transport defects may result in a metabolic crisis in the immediate newborn period before feeding is initiated, as in the infantile forms of carnitine palmitoyltransferase two deficiency and carnitine-acylcarnitine translocase deficiency (Chalmers et al. 1997). Many cases of acute fatty liver of pregnancy are a manifestation of recessively inherited FAO defects in the fetus, the most common being mitochondrial trifunctional protein defect that impairs oxidation of long-chain fatty acids (Ibdah et al. 1999; Yang et al. 2002).
Several FAO defects, including medium-chain acyl-CoA dehydrogenase, long-chain acyl-CoA dehydrogenase, carnitine transporter defect, and carnitine translocase defect, have been implicated in a small number of sudden deaths in infancy and childhood.
13.6.2 Pathology
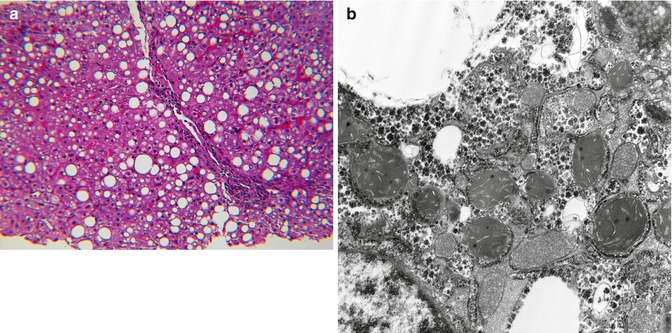
Based upon observations in liver biopsies and autopsy material, infants and children with defective FAO accumulate neutral lipid in organs most dependent upon FAO such as the liver, heart, proximal renal tubules, and type 1 skeletal muscle fibers. The lipid accumulation occurs predominantly in the form of microvesicular steatosis in which the lipid vacuoles indent but do not displace the nucleus (Fig. 13.9a, b); coexistent large droplet lipid is usually associated, but often a lesser component. In MCAD, the accumulation of lipid in hepatocytes may be transient and may abate or disappear without permanent liver injury when homeostasis is restored. Analysis of urine for acylcarnitine compounds and establishment of a fibroblast culture are essential supplements to appropriate tissue samples when clinical suspicion is high. Limited published observations on antemortem liver samples suggest that ultrastructure of mitochondria in FAO are not distinctive in comparison to mitochondrial alterations in primary disorders of electron transport, mitochondrial DNA (mDNA) depletion, and epidemic Reye Syndrome.
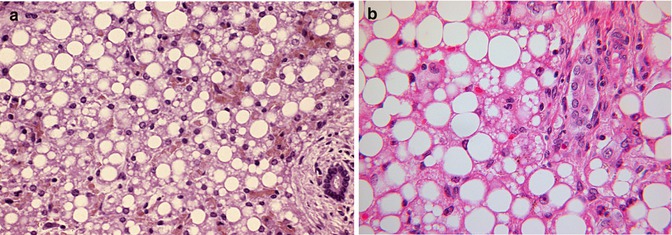
Fig. 13.9
(a) Liver in MCAD with both micro- and macrovesicular steatosis. H&E stain. (b) Liver in LCAD with both micro- and macrovesicular steatosis. H&E stain
13.6.3 Diagnosis
Diagnosis is usually based upon a characteristic abnormal urine acylcarnitine profile determined by high-performance liquid chromatography (HPLC) and plasma acylcarnitine profile determined by fast atom bombardment mass spectrometry from Guthrie card bloodspots, organic acid profiles, and studies of fatty acid processing performed on cultured fibroblasts. Mutation analysis is available for the most common mutations in MCAD.
13.7 Mitochondriopathies
13.7.1 Clinical Manifestations
Mitochondriopathies are abnormalities of energy metabolism caused either by nuclear DNA mutations or mtDNA mutations/deletions that impair oxidative phosphorylation (OXPHOS) (Finsterer 2004; Dimauro 2011). Clinical manifestations in infants or children may involve one or more organs, often in clinically recognizable patterns, but the spectrum differs somewhat from those that affect adults (Bernier et al. 2002). Though enigmas remain, understanding continues to evolve as knowledge and experience expand and the difference between primary and secondary mitochondrial dysfunction due to other metabolic disturbances is clarified.
Mitochondriopathies first recognized, such as those characterized by ragged-red muscle fibers, usually do not affect the liver. Most are caused by maternally inherited mtDNA mutations that typically affect one particular component of OXPHOS system. Clinical manifestations are usually delayed until the proportion of abnormal mitochondria in a given organ, usually skeletal muscle, exceeds a critical threshold that varies with metabolic demand. Large deletions in mtDNA cause Kearns-Sayre syndrome, a myopathy in which the liver is rarely involved, and Pearson marrow-pancreas syndrome. Pearson syndrome, the more common of the two in infants and young children, causes anemia, both exocrine and endocrine pancreas dysfunction, and liver disease that may be clinically significant (Williams et al. 2012). Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a multisystem disease that affects young adults but may begin in childhood; the liver is not usually involved (Garone et al. 2011).
Nuclear DNA (nDNA) mutations may affect one particular enzyme in the Krebs citric acid cycle, one subunit of the OXPHOS system, or generally impair mtDNA synthesis resulting in multiple defects in OXPHOS. The liver may be affected alone or in various combinations with brain, heart, kidney, skeletal muscle, or gastrointestinal tract.
Acute liver failure due to mitochondriopathy is rare and usually manifest in infants, many of whom have or will develop significant brain disease (Sarzi et al. 2007). One cause, deficiency of cytochrome oxidase (complex IV), associates with many phenotypes, one of which is congenital lactic acidosis, a systemic disease with devastating clinical manifestations that suggest multiorgan dysfunction. Another phenotype, GRACILE syndrome (intrauterine growth restriction, aminoaciduria, cholestasis, hepatic iron overload, and lactic acidosis), is due to mutation in BCS1L, a chaperon protein for complex III, that results in reduced levels of this electron transporter. Still another phenotype, Wolcott-Rallison syndrome, consists of neonatal-infantile diabetes mellitus, skeletal dysplasia, growth retardation, and intermittent liver failure. In these infants, complex I is deficient due to mutation in a nuclear gene, PRK-like ER kinase. Recent studies show that infant liver failure is often caused by another mechanism, mtDNA depletion, that results from mutations in nuclear genes that control synthesis of most subunits of the OXPHOS system (Fellman and Kotarsky 2011). Examples of mutations in nuclear genes or nuclear-coded proteins that are reported to cause progressive hepatic or hepatocerebral mitochondriopathy in infancy are deoxyguanosine kinase, POLG1, SUCLG1, and MPV17. The phenotypic spectrum of MPV17 mutation-related disease includes the Navajo neurohepatopathy (Holve et al. 1999; Karadimas et al. 2006).
Most, if not all, of the valproic acid-associated cases of acute liver failure in infants or children with seizures have an underlying mitochondriopathy that affects both liver and brain. Such patients are exceptionally vulnerable to valproic acid because it interferes with mitochondrial oxidation of fatty acids (Silva et al. 2008). Most cases of Alpers-Huttenlocher syndrome are mitochondriopathies in which brain involvement leads to early presentation as a seizure disorder (Gordon 2006). The well-known association of progressive liver and brain mitochondriopathy is the basis for the warning to avoid liver transplants in infants or children with liver failure unless brain involvement can reasonably be excluded.
Mitochondrial hepatotoxicity of nucleoside reverse transcriptase inhibitors of DNA polymerase gamma may cause steatosis and lactic acidemia. These complications of therapy for chronic viral infections are rare in children perhaps because long-term exposure is necessary, but have been reported in infants exposed during pregnancy.
13.7.2 Pathology
Liver histological features at the time of liver failure due to a primary mitochondriopathy vary depending on the age of onset and stage of the disorder. Typical changes in older infants at the time of liver failure include patchy, sometimes extensive micro- and macrovesicular steatosis, intralobular cholestasis, finely granular bright red hepatocytes isolated or clumped in groups that contain excessive numbers of mitochondria, swollen hepatocytes that lack lipid droplets, scattered necrotic hepatocytes, foci of intralobular regeneration and collapse, and mixed portal/lobular inflammation accompanied by progressive portal fibrosis (Fig. 13.10a–d). Oxidative enzyme histochemistry applied to cryostat sections of the liver may be helpful in recognition of mitochondriopathy. Selective reduction or hyperintensity of histochemical reactions for cytochrome oxidase and succinic dehydrogenase, normally uniform in cryostat sections of liver, may be observed (Ibdah et al. 1999). In the liver, as well as in organs such as the heart, mitochondriopathy may induce generalized proliferation of mitochondria despite functional deficiency. The generic histological phenotype described above may be absent in fulminant mitochondriopathies that cause lethal lactic acidosis in the newborn period (Fig. 13.10e). Immunohistochemical stains may be useful to demonstrate mitochondrial hyperplasia.
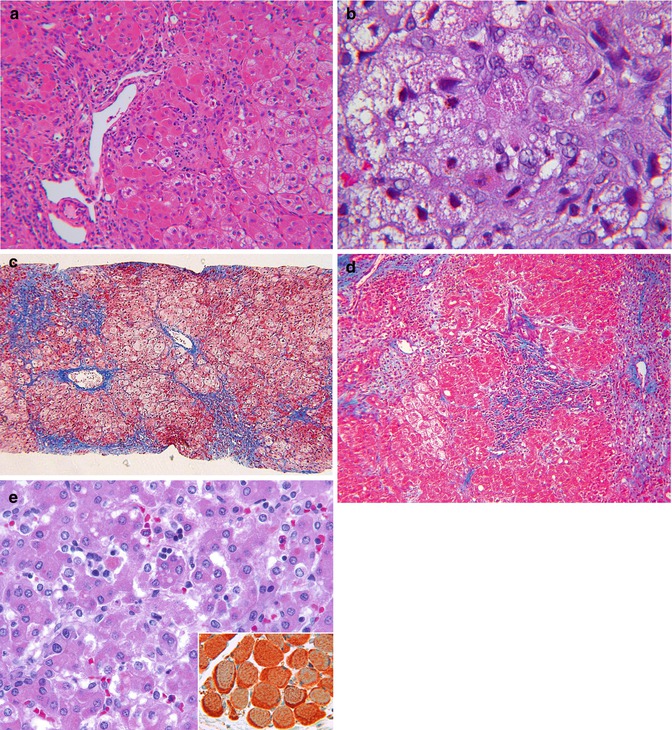
Fig. 13.10
(a) Mitochondriopathy. mDNA depletion in late infancy. Patches of eosinophilic faintly granular hepatocytes are intermixed with swollen hepatocytes. Lipid is uncommon. Portal and lobular inflammation are mild. H&E stain. (b) Mitochondriopathy, MPV17 defect. Microvesicular steatosis is prevalent. Two isolated granular red hepatocytes contain excess mitochondria. H&E stain. (c) Mitochondriopathy, MPV17 defect with swollen hepatocytes and minimal steatosis. Note periportal fibrosis with early bridging and pericellular fibrosis. Trichrome stain. (d) Mitochondriopathy, valproate-induced acute liver failure. Periportal fibrosis is flanked by areas of zone 3 collapse. Trichrome stain. (e) Congenital lactic acidosis. Universal fine granular hepatocyte cytoplasm is due to mitochondrial hyperplasia, present also in heart and skeletal muscle. Lipid vacuoles are a minor feature. H&E stain. Inset: massive accumulation of mitochondria in diaphragm muscle verified by immunohistochemical stain
Distinctive ultrastructural mitochondrial abnormalities are reported in many infants and young children with proven mitochondriopathy (Mandel et al. 2001; Labarthe et al. 2005; Wong et al. 2007). These include pleomorphism of size and shape, increased or decreased numbers of mitochondria per hepatocyte, bizarre dilatation of cristae, and expansion of matrix that displaces cristae as it accumulates causing mitochondrial enlargement that may be spectacular. Matrix may be pale or dense (Fig. 13.11a–d). Similar morphological changes occur in acute liver failure associated with what formerly were called “idiosyncratic” drug reactions to valproic acid used to treat seizures in infants or in older children. Many of these patients have an unrecognized genetic mitochondriopathy that causes mDNA depletion. Curiously, intra-cristal paracrystalline inclusions of the type that are common in the ragged-red fibers of mitochondrial myopathy are rarely, if ever seen in mitochondrial hepatopathy. Whether mitochondrial ultrastructural changes may be specific for particular defects requires more observation.
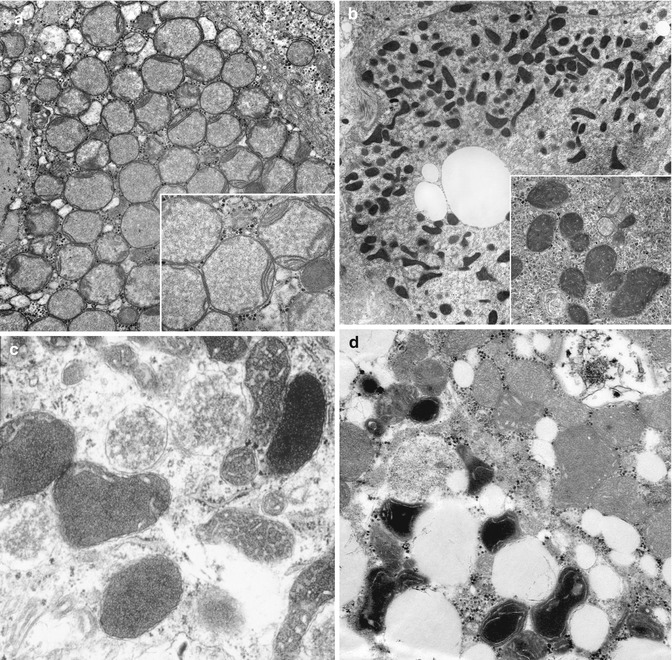
Fig. 13.11
(a) Mitochondriopathy in subacute liver failure in late infancy due to mDNA depletion. Mitochondrial matrix is pale, flocculant, and expanded and displaces cristae to periphery. EM. (b) Mitochondrial pleomorphism in multiacyl-COA dehydrogenase deficiency. Inset: pleomorphic mitochondria contain dilated cristae, coarsely granular matrix, and increased dense granules. EM. (c) Mitochondriopathy in valproate-induced liver failure. Mitochondrial pleomorphism, variable accumulation of dense matrix and dilatation of cristae. EM. (d) Mitochondriopathy in acute liver failure due to MPV17 defect and mDNA depletion. Wide range of mitochondrial abnormality includes pleomorphism, variable accumulation of abnormally dense matrix, and dilatation of cristae. EM
The range of ultrastructural changes in primary mitochondriopathy appears to differ from changes due to nonspecific oxidative metabolic stress that accompany ethanolism, nonalcoholic steatohepatitis, urea cycle disorders, bile acid synthesis defects, extrahepatic portal vein obstruction, and rarely in glycogenosis. These changes include normal mitochondria with mild pleomorphism, scattered pale swollen mitochondria, paracrystalline matrix inclusions, and megamitochondria (Figs. 13.7 and 13.14d).
13.7.3 Diagnosis
Criteria for clinical and laboratory diagnosis of mitochondriopathy in the context of neuromuscular disease with or without multiorgan involvement are not perfect (Gordon 2006). These criteria are even less helpful when mitochondrial disease primarily affects one or more visceral organs such as the liver. Skeletal muscle is commonly affected in multiorgan mitochondriopathies but does not always exhibit diagnostic changes by light or electron microscopy. Nonetheless, muscle biopsy is convenient and, properly triaged and processed, has a reasonably high yield as a screening test for biochemical and molecular genetic disorders (nDNA or mtDNA). Muscle biopsy may be useful when clinical signs indicate primary liver involvement alone or in conjunction with central nervous system disease.
Light and electron microscopic findings in needle biopsy samples of the liver often create suspicion for mitochondrial hepatopathy and may occasionally be diagnostic though not specific for a particular genetic entity. mtDNA content may be assessed in a needle biopsy specimen. Specific mutations in nDNA that cause mtDNA depletion and liver failure in infants or young children can now be assayed in blood leukocyte DNA. However, open liver biopsy may be needed for a complete assessment that includes light and electron microscopy, measurement of electron transport activities, mDNA content, and search for mDNA deletions or point mutations. With the advent of mitoexome and whole genome sequencing, DNA from the liver, cultured fibroblasts and leukocytes will modify the role of liver biopsy.
13.7.4 Reye Syndrome (RS)
RS is an acute liver dysfunction with encephalopathy and fatty degeneration of the liver. RS occurs in two clinical guises. One form affects older, previously well children, peaked between 1970 and 1985 and has almost disappeared. The other form mainly affects infants and young children, as a complication of an underlying metabolic disorder. The former was a transient post-viral acute encephalopathy associated with fatty degeneration of hepatocytes in which microvesicular steatosis developed in the wake of a viral illness along with signs of brief transient failure of hepatic synthetic function, hyperammonemia, and mild to moderate elevation of serum aminotransferases. Clear evidence of association of RS with epidemics of chicken pox and influenza B was followed by epidemiological evidence linking the syndrome to low-therapeutic levels of salicylate. Mortality and long-term morbidity due to brain injury was about 5 %. Both light and electron microscopy of liver tissue obtained early in the course of the disease were distinctive and established the basis as a hyperacute emergence (phanerosis) of microvesicular fat as a marker for acute liver failure due to a transient reversible mitochondriopathy (Partin et al. 1971; Bove et al. 1975). Concomitant glycogen depletion was often severe, accounting for hypoglycemia. The hepatocytes in the early phase are swollen and contained few visible lipid droplets in paraffin sections (Fig. 13.12). However, abundant microvesicular lipid, apparently concealed during rapid evolution of the hepatopathy, was demonstrable in fat stains on frozen sections. Zonal necrosis was absent, but apoptosis was often seen. Histochemical studies showed depletion of succinic dehydrogenase and cytochrome oxidase. Ultrastructural changes in mitochondria were distinctive and remain so in retrospect, suggesting kinship to the “membrane permeability transition” pathway. The biology of the relationship of RS to low blood levels of salicylate remains mysterious, and the validity of the statistical link, though generally accepted, has been questioned.
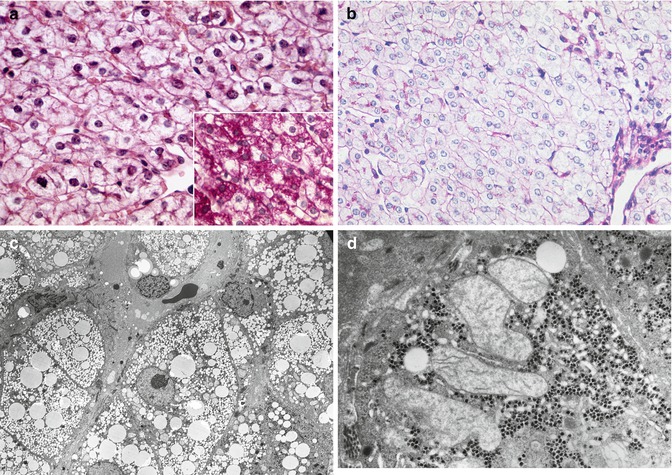
Fig. 13.12
(a) Reye syndrome. Transient encephalopathy. Swollen hepatocytes without obvious vacuolation contain concealed lipid. H&E stain. Inset: mild glycogen depletion. PAS stain. (b) Reye syndrome. Severe encephalopathy. Swollen hepatocytes contain massive amounts of concealed lipid and no stainable glycogen. PAS stain. (c) Reye syndrome, with abundant microvesicular lipid visible in resin-embedded section. Toluidine blue-Azure blue stain. (d) Reye syndrome, enlarged swollen ameboid mitochondria with pale watery matrix and displaced cristae coexist with preserved glycogen. EM
With decline of aspirin usage as an antipyretic in children, RS has almost, but not completely, disappeared in the United States (Belay et al. 1999) except in the context of acute decompensation of an underlying metabolic disorder of energy metabolism typically in infants or very young children. In such cases, the distinctive ultrastructural changes of mitochondria in epidemic RS are absent (Garone et al. 2011). Skepticism about the existence of RS except in the context of a specific metabolic disorder is based upon advances over the past 30 years in diagnostic techniques that were not available at the height of the epidemic. However, this argument is unconvincing given the prevalence of epidemic RS in older children and rarity of second episodes in the vast majority who survived.
13.7.5 Wilson Disease (WD)
WD is an autosomal recessive disorder of copper transport due to mutations in the ATP7B gene that results in copper overload in the liver, progressive liver disease, and degeneration of central brain nuclei often with onset in childhood. Laboratory criteria for diagnosis are low serum ceruloplasmin, elevated 24-h urine copper, and high hepatic tissue copper levels, but may not be definitive (Nicastro et al. 2010). Early signs in children such as elevated serum transaminases and/or hepatomegaly or acute onset of liver failure may prompt an investigation that includes liver biopsy. Liver histology ranges from normal to chronic hepatitis with or without steatosis, often prominent nuclear glycogenation, or submassive necrosis. The rhodanine copper stain identifies copper within secondary lysosomes and is widely used but not specific, because copper accumulates nonuniformly in WD and may also be detected in other conditions, particularly if cholestasis is a confounding factor. Liver ultrastructure may be helpful showing prominent mitochondrial changes such as tubular dilatation of cristae and matrix inclusions, as well as unusually complex membrane-bound insoluble lipid inclusions, dubbed lipolysosomes (Fig. 13.13a, b). The mitochondrial changes in Wilson disease overlap with those seen in primary mitochondriopathies and also with those seen in acquired conditions such as steatohepatitis where oxidative stress may be causative. Evidence from humans and animal models of genetic and acquired copper toxicity suggests that copper localizes in and promotes oxidant injury to mitochondria thereby playing an essential role in progressive liver and brain damage in Wilson disease (Sokol et al. 1994; Zischa et al. 2011).
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
