Small Intestine
Although the small intestine comprises 75% of the length and 90% of the surface area of the alimentary tract, neoplasms of the small intestine are rare (accounting for only 1% to 2% of all gastrointestinal (GI) neoplasms, and <1% of all cancers in the United States). In daily practice, small intestinal neoplasms are seldom seen in mucosal biopsy material and, sadly, are often clinically occult until they attain untreatable dimensions and stages. There are essentially four types of primary malignant neoplasms that affect the small intestine. In descending frequency, they are adenocarcinomas, well-differentiated neuroendocrine tumors (WDNTs; carcinoids), lymphomas, and sarcomas (1). Malignant neoplasms detected in the small intestine are often metastases. When interpreting small intestinal biopsies, be alert to these features:
(a) Reactive changes overlying neoplasms that are too deep to be detected on biopsy material can easily mimic Crohn disease, especially those found with WDNTs (carcinoids).
(b) The superficial portions of WDNTs may be quite subtle (Figs. 3.1 and 3.2, e-Figs. 3.1-3.5). Unlike indolent gastric WDNTs associated with atrophic gastritis, those in the small intestine are generally not associated with inflammatory conditions.
(c) Metastatic carcinoma from many sites can mimic an intraepithelial component when it extends along the surface of the small intestine (2).
(d) Pancreatic lesions can spread directly onto the ampullary surface, thus mimicking primary small bowel lesions (e-Figs. 3.6 and 3.7).
(e) Always scan dilated lacteals for isolated neoplastic cells (e-Figs. 3.8 and 3.9). Common primaries that metastasize to the duodenum include breast, lung, and melanoma. Interestingly, mesothelioma can spread to the small bowel, either directly (peritoneal lesions) or as metastases from the pleura (e-Figs. 3.10-3.14) (3-6).
(f) Prominent crushed Brunner glands can be mistaken for neural lesions. This pitfall is easily avoided by ordering a PAS stain in doubtful cases (Fig. 3.3, e-Figs. 3.15 and 3.16).
(g) The duodenum, including the ampullary region, is prone to reparative changes that can mimic adenomas.
 FIGURE 3.1 Well differentiated neuroendocrine tumor (WDNT). Superficial portions of WDNTs can be quite subtle, as in this example. The neoplastic cells are seen here within the lamina propria on the right-hand side of the field. |
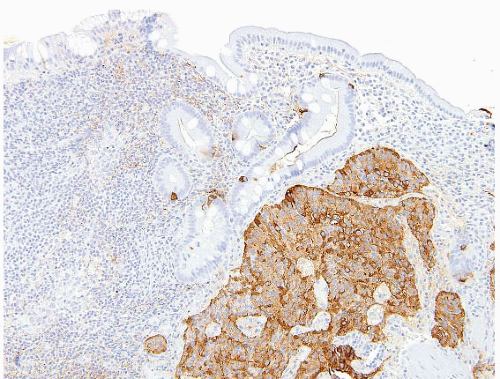 FIGURE 3.2 Well differentiated neuroendocrine tumor (WDNT). A chromogranin stain highlights the neoplastic cells in this example of a WDNT. |
POLYPS
Adenomas
Small intestinal adenomas are usually found in the duodenum, and like their colon counterparts, have three major histologic types: tubular, tubulovillous, and villous. Though rare and not reported in the literature as of this writing, we have encountered one traditional serrated adenoma of the duodenum (e-Figs. 3.17-3.20). In patients with familial adenomatous polyposis (FAP), endocrine cells are often a prominent component (e-Fig. 3.21). Though classically described in the stomach, pyloric gland adenomas can also be encountered in the duodenum (Fig. 3.4, e-Figs. 3.22-3.28) (7,8). Most adenomas occur singly; the presence of multiple adenomas in the small intestine is unusual in the absence of FAP (Fig. 3.5, e-Figs. 3.29 and 3.30). In FAP patients, after colectomy, the main cause of death is upper GI tract malignancy (9). The majority of FAP patients also develop upper GI tract polyps; those in the gastric antrum and duodenum are usually neoplastic. One study documented the occurrence of ileal pouch adenomas in 22.8% (8/35) of FAP patients who underwent proctocolectomies. Eight of those patients also had jejunal and ileal adenomas diagnosed by capsule endoscopy (10). Therefore, surveillance of FAP patients with upper endoscopy and/or wireless capsule endoscopy with biopsies is recommended (9,11). Because of their potential to undergo malignant transformation, adenomas should be removed via endoscopic polypectomy for pedunculated tumors and endoscopic mucosal resection, or surgical resection, for large sessile lesions. Many
surgical colleagues manage high-grade dysplasia in these lesions with radical surgery, so the pathologist should have a high threshold for diagnosing high-grade dysplasia in these lesions (e-Fig. 3.31).
surgical colleagues manage high-grade dysplasia in these lesions with radical surgery, so the pathologist should have a high threshold for diagnosing high-grade dysplasia in these lesions (e-Fig. 3.31).
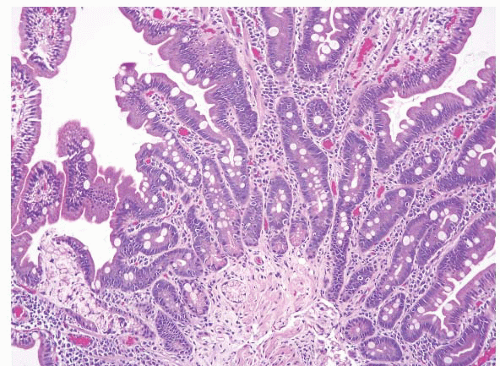 FIGURE 3.3 Crushed Brunner glands. Crushed Brunner glands are seen here (lower left-hand side of the field) within a crypt. A PAS stain will highlight these glands bright pink (see e-Fig. 3.16). |
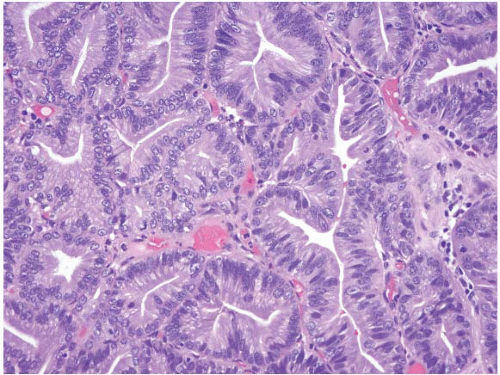 FIGURE 3.4 Pyloric gland adenoma. These polyps show tightly packed neoplastic pyloric-type glands with grayish, ground-glass cytoplasm and round-to-oval small nuclei with small nucleoli. |
 FIGURE 3.5 Duodenal tubular adenomas. Multiple duodenal tubular adenomas associated with an invasive adenocarcinoma (right-hand side of the field) are seen here in a patient with Familial adenomatous polyposis (FAP). |
Probably the biggest pitfall in interpreting adenomas in the small bowel is the proclivity of reparative lesions to mimic them. When peptic duodenitis has a nodular configuration, together with marked reactive epithelial changes on microscopic exam, it is easily confused with an adenoma (Fig. 3.6, e-Fig. 3.32). One clue to recognizing peptic duodenitis
is that it often has surface gastric mucin cell metaplasia in the atypical focus. Also, adenomas have their initiation point just beneath the surface epithelium (12), whereas reparative lesions that occur with peptic duodenitis have theirs at the base of the mucosa. In addition, the surface cells of duodenal adenomas tend to have lipid accumulation in the neoplastic enterocytes. Presumably, since they are neoplastic, these cells can absorb lipid but then are unable to package it for further digestion so that there is often prominent “lipid hangup” in the surface of duodenal adenomas (Figs. 3.7 and 3.8, e-Figs. 3.33 and 3.34). This is nicely highlighted on PAS/AB stains since fat does not take up the PAS stain.
is that it often has surface gastric mucin cell metaplasia in the atypical focus. Also, adenomas have their initiation point just beneath the surface epithelium (12), whereas reparative lesions that occur with peptic duodenitis have theirs at the base of the mucosa. In addition, the surface cells of duodenal adenomas tend to have lipid accumulation in the neoplastic enterocytes. Presumably, since they are neoplastic, these cells can absorb lipid but then are unable to package it for further digestion so that there is often prominent “lipid hangup” in the surface of duodenal adenomas (Figs. 3.7 and 3.8, e-Figs. 3.33 and 3.34). This is nicely highlighted on PAS/AB stains since fat does not take up the PAS stain.
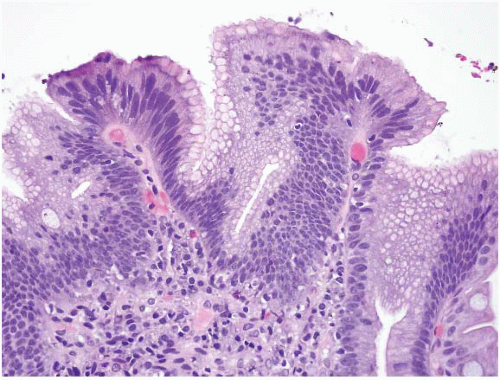 FIGURE 3.6 Nodular chronic peptic duodenitis. When peptic duodenitis has a nodular configuration, together with marked reactive epithelial changes on microscopic exam, it is easily confused with an adenoma. Although there is nuclear enlargement and hyperchromasia, the presence of foveolar cell metaplasia is a clue that helps to interpret the changes as reactive rather than neoplastic. |
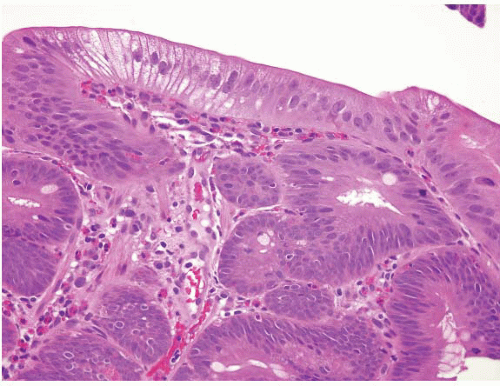 FIGURE 3.7 Duodenal adenoma. Epithelial cells from true adenomas can absorb lipid but are unable to package it for further digestion such that there is often prominent “lipid hangup” at the surface, as in this example (see Fig. 3.8). |
Unfortunately, there are occasional cases in which it is impossible to distinguish adenomas from reparative processes and we designate them as “indefinite for dysplasia” and request additional samples (e-Figs. 3.35 and 3.36). This distinction is not trivial in the small intestine, particularly in the area of the ampulla. There is a precedent for treating large ampullary adenomas with pancreatoduodenectomy, based on their high likelihood of harboring an occult invasive carcinoma (13). More recently, however, endoscopic papillectomy has been increasingly advocated with some success as an alternative to surgery (14).
The ampulla itself is often a source of difficulty since it is normal to have ampullary glands, with their features akin to biliary epithelium, interspersed with disorganized bundles of smooth muscle (e-Figs. 3.37-3.42). When inflammation is a feature, great caution is advised. The ampulla is not typically biopsied without a compelling reason because pancreatitis may be a severe consequence of performing such biopsies.
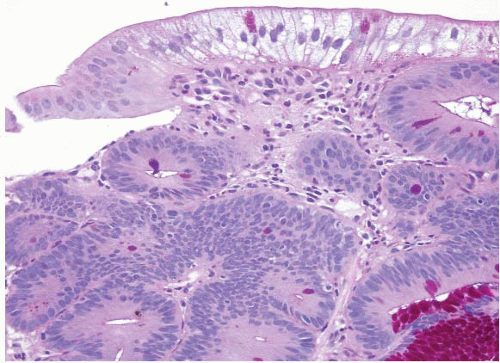 FIGURE 3.8 Duodenal adenoma. Intracytoplasmic lipid appears clear in this PAS preparation of a duodenal tubular adenoma. |
When a clearly neoplastic small bowel lesion displays peculiar features, it is always important to consider if the process is a metastasis or if it extends directly from the pancreatobiliary tree.
Peutz-Jeghers Polyps
The World Health Organization (WHO) has defined Peutz-Jeghers (PJ) syndrome as an inherited cancer syndrome characterized by mucocutaneous melanin pigmentation and hamartomatous intestinal polyposis, preferentially affecting the small intestine (15). The most frequently reported malignancy in these patients is colorectal cancer followed by breast, small bowel, stomach, and pancreas cancer in descending order of frequency. The cumulative GI cancer risk at age 60 to 70 years is between 38% and 66%, as reported in a recent systematic review of the literature (16) and the cumulative lifetime risk of cancer in such patients has been estimated at 93% in one study (17). Other extraintestinal neoplasms include those of the endometrium, lung, ovary (sex cord tumors with annular tubules), cervix (adenoma malignum), testis (sertoli cell tumors), pancreas (carcinomas), and breast (carcinomas). The average age of developing malignancy in patients with PJ syndrome is 42 years (16). This syndrome is about one tenth as common as FAP (the incidence of FAP is between 1:7,000 and 1:30,000).
The diagnostic criteria include any or all of the following features: (a) three or more histologically confirmed PJ polyps; (b) any number of PJ polyps in an individual with a family history of this syndrome; (c) prominent mucocutaneous melanin pigmentation in an individual with affected relatives; or (d) any number of polyps in an individual with prominent mucocutaneous pigmentation. Mucocutaneous pigmentation should be conspicuous as some individuals without the syndrome may have some degree of pigmentation. Conversely, this pigmentation may regress to involve only the inside of the mouth with increasing age.
In addition, some data suggest that having even a single small intestinal PJ polyp confers the same risk as having the full syndrome (18). Recognizing the syndrome is important, based on the associated 10- to 18-fold increase in cancers of both the intestines and extraintestinal sites. This syndrome is an autosomal dominant one with virtually 100% penetrance. Mutations or deletions in an involved gene, LKB/STK11, are found in 80% to 94% of affected patients (19,20).
In their classical appearance, PJ polyps are easy to recognize on endoscopic biopsies. They are hamartomas and thus display the type of mucosa typical for the site in which they are found. Thus, in the stomach they have gastric mucosa, and in the small bowel they have small intestinal mucosa. The problems arise when biopsies are superficial or when ulceration distorts them. However, PJ polyps in the small intestine are far more likely than those in the stomach to display the classic arborizing, smooth muscle cores from which the mucosa leafs out (Figs. 3.9 and 3.10, e-Figs. 3.43-3.48). Although these polyps may have associated
dysplasia (e-Figs. 3.49 and 3.50) or (rarely) invasive carcinoma (21), most GI malignancies associated with this syndrome seldom arise from these lesions.
dysplasia (e-Figs. 3.49 and 3.50) or (rarely) invasive carcinoma (21), most GI malignancies associated with this syndrome seldom arise from these lesions.
What is the clinical significance of encountering an isolated PJ polyp in a patient with no clinical manifestations of PJ syndrome? Burkart et al. addressed this question in a study that included eight patients with “isolated” PJ polyps. Three of these polyps arose in the small bowel and showed unequivocal PJ polyp histology. The remaining five patients had colonic polyps with features suggestive but not fully diagnostic of PJ polyp. Interestingly, all three patients with unequivocal PJ polyps had clinical
histories suggestive of PJ syndrome (two had pancreatic cancer, one had bilateral ovarian masses and glomus tympanicum tumor) but did not fulfill the WHO diagnostic criteria. The remaining five patients with equivocal colonic polyps had personal or family history of neoplasia, save for one individual. These findings suggest that patients with a solitary PJ polyp are at risk for PJS-associated cancers and that if solitary PJ polyps exist, they are extremely rare (18).
histories suggestive of PJ syndrome (two had pancreatic cancer, one had bilateral ovarian masses and glomus tympanicum tumor) but did not fulfill the WHO diagnostic criteria. The remaining five patients with equivocal colonic polyps had personal or family history of neoplasia, save for one individual. These findings suggest that patients with a solitary PJ polyp are at risk for PJS-associated cancers and that if solitary PJ polyps exist, they are extremely rare (18).
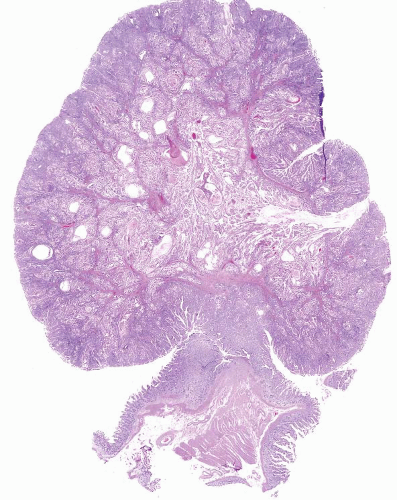 FIGURE 3.9 Small bowel Peutz-Jeghers (PJ) polyp. Small bowel PJ polyp is shown here. Small intestinal examples are far more likely than those in the stomach to display the classic arborizing, smooth muscle cores from which the mucosa leafs out, as seen in this example. |
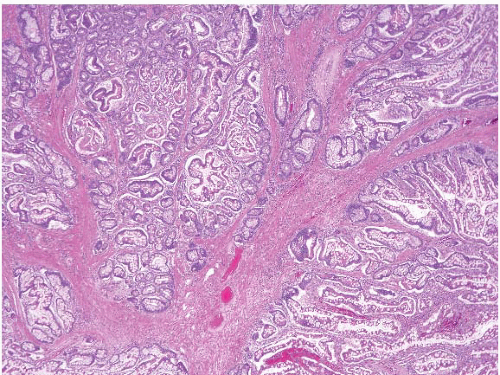 FIGURE 3.10 Small bowel Peutz-Jeghers (PJ) polyp. This higher magnification image of Figure 3.9 shows thick bundles of smooth muscle surrounding groups of glands in this classic example of a PJ polyp. |
Juvenile Polyps
Juvenile polyposis is a genetically heterogeneous condition in which some families have autosomal dominant germline mutations in the DPC4/SMAD4 gene on chromosome 18q21. The polyps in juvenile polyposis can be limited to the colon or can be generalized, involving the colon, small bowel, and stomach. Though there are reports of patients with gastricrestricted juvenile polyposis, we are unaware of isolated syndromic small bowel examples. Nevertheless, small bowel juvenile polyps can rarely be encountered and show similar morphologic features of those arising elsewhere in the GI tract with rounded, sometimes eroded surface, dilated glands, and edematous and inflamed lamina propria (Figs. 3.11 and 3.12, e-Figs. 3.51-3.54). These polyps appear virtually identical to Cronkhite-Canada polyps (see the following section), but the flat mucosa between juvenile polyps is normal in contrast to the abnormal mucosa between Cronkhite-Canada polyps. Juvenile polyposis is further discussed in Chapters 2 and 4.
 FIGURE 3.11 Small intestine juvenile polyp. Juvenile polyps show an edematous and inflamed lamina propria along with dilated glands. |
Cronkhite-Canada Polyps
Cronkhite-Canada is a rare hamartomatous polyposis syndrome initially described in 1955 by Leonard W. Cronkhite and Wilma J. Canada (22). Most cases have been reported in Japan. Patients with this condition
have diffuse polyposis throughout the GI tract (except the esophagus) associated with alopecia, skin hyperpigmentation, nail atrophy, protein losing enteropathy, vitamin deficiencies, and electrolyte imbalances. Patients present with taste disturbances, diarrhea, weight loss, abdominal pain, anemia, and peripheral edema. No associated germline mutations or familiar predisposition have been identified to date. Mortality is high and severe GI bleeding, intussusception, malnutrition, infection, and rectal prolapse may complicate the clinical course. Cases associated with myelodysplastic syndrome and membranous glomerulonephritis have been reported (23-25).
have diffuse polyposis throughout the GI tract (except the esophagus) associated with alopecia, skin hyperpigmentation, nail atrophy, protein losing enteropathy, vitamin deficiencies, and electrolyte imbalances. Patients present with taste disturbances, diarrhea, weight loss, abdominal pain, anemia, and peripheral edema. No associated germline mutations or familiar predisposition have been identified to date. Mortality is high and severe GI bleeding, intussusception, malnutrition, infection, and rectal prolapse may complicate the clinical course. Cases associated with myelodysplastic syndrome and membranous glomerulonephritis have been reported (23-25).
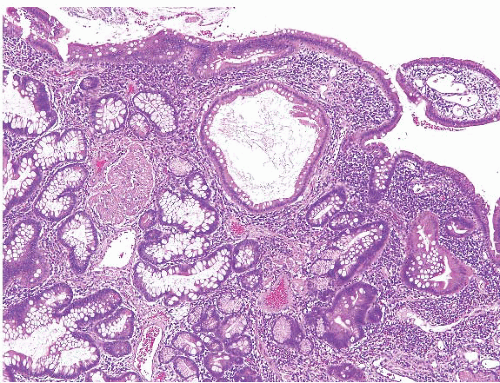 FIGURE 3.12 Small intestine juvenile polyp. This higher magnification image of Figure 3.11 better shows the expanded lamina propria and dilated gland of a small intestinal juvenile polyp. |
Microscopic exam shows a polyp with a broad sessile base, expanded edematous lamina propria, and cystic glands (Fig. 3.13, e-Fig. 3.55). Distinguishing between Cronkhite-Canada and juvenile polyps is essentially impossible while unaware of the clinical and endoscopic findings. If the endoscopist has biopsied the flat mucosa between the polyps, the flat mucosa is normal in patients with juvenile polyposis, whereas it is abnormal in Cronkhite-Canada syndrome. Finding dysplastic changes favors juvenile polyposis as dysplasia is essentially never seen in Cronkhite-Canada polyps, but dysplasia occasionally complicates juvenile polyps.
Whether polyps in Cronkhite-Canada syndrome possess malignant potential remains a matter of debate. Though there are reports in the literature of cases associated with adenomas and carcinoma, it is difficult to
determine whether this association is significant or merely coincidental (24-27). Nutritional support and oral steroid administration provide symptomatic relief in some patients (23,26,27). See additional discussion of Cronkhite-Canada syndrome and illustrations in Chapters 2 and 4.
determine whether this association is significant or merely coincidental (24-27). Nutritional support and oral steroid administration provide symptomatic relief in some patients (23,26,27). See additional discussion of Cronkhite-Canada syndrome and illustrations in Chapters 2 and 4.
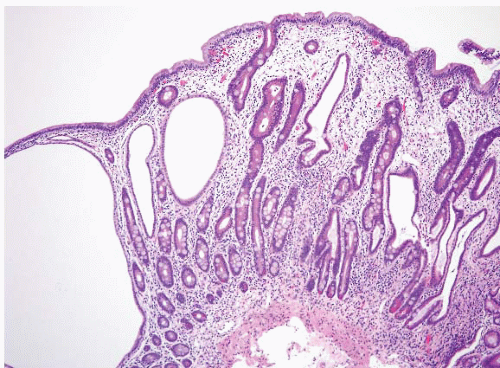 FIGURE 3.13 Cronkhite-Canada polyp. Polyps arising in the setting of Cronkhite-Canada syndrome can be histologically identical to juvenile polyps. They have a broad, sessile base, expanded edematous lamina propria, and cystically dilated glands. A diagnostic clue is that the surrounding flat mucosa is abnormal. |
Brunner Gland Polyps
Brunner glands are unique to the duodenum and typically reside in the submucosa. Their ducts communicate to the lumen opening within the crypts between the intestinal villi. They produce alkaline mucus that neutralizes acidic luminal contents protecting the mucosa from autodigestion. They are usually found in tightly packed aggregates and have abundant grey, PAS-positive cytoplasm and small, basally located nuclei. Small Brunner gland lesions are commonly encountered in daily practice, mostly as part of the reactive changes seen in the setting of chronic peptic duodenitis (CPD). In CPD, Brunner glands can be seen traversing the muscularis mucosae and extending into the lamina propria (Fig. 3.3, e-Figs. 3.15 and 3.16). If sufficient glands accumulate within the lamina propria this may produce the endoscopic appearance of a nodule and the term “Brunner gland hyperplasia” may be used in this setting. When a nodule is seen endoscopically and we see intramucosal Brunner glands in association with the changes of CPD (gastric mucin cell metaplasia, reactive epithelial changes), we diagnose “nodular chronic peptic duodenitis”. A literature search discloses no well-documented cases of Brunner gland-associated dysplasia or carcinoma and, as a result, we avoid the term “Brunner gland adenoma.”
“Brunner gland hamartoma” refers to a pedunculated polyp composed of Brunner glands and surrounding fibrous stroma that usually measures more than 2 cm. Patients may present with intussusception, hemorrhage, and/or obstruction. Lesions can attain large sizes, with one lesion reported in the literature measuring 10 cm (28). Hur et al. report on the radiologic and pathologic findings of nine Brunner gland hamartomas (29). The authors describe macroscopically well-defined, lobulated solid masses with intervening fibrous septa and areas of cystic change. Histologically one sees a submucosal-based mass with an overlying intact mucosa composed of nests of benign Brunner glands separated by smooth muscle and vascular septa associated with areas of cystic change (Figs. 3.14 and 3.15, e-Fig. 3.56). Areas of adipose tissue and chronic inflammation may be seen. Hur et al. comment on two cases with high-grade dysplasia in their study, a finding that likely reflects a lower “dysplasia threshold” among our Eastern colleagues.
On a practical note, we often designate smaller such lesions as “Brunner gland polyp” as there are no data that prove whether these lesions are reactive, hamartomatous, or neoplastic, although some examples display features that suggest that they are indeed adenomas.
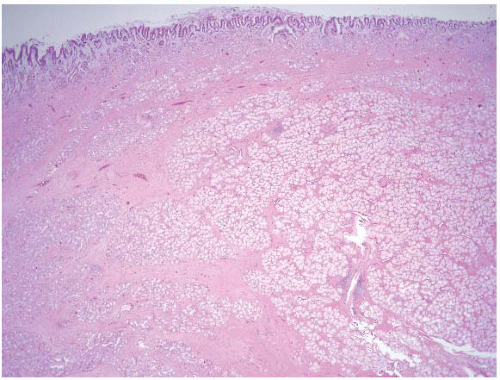 FIGURE 3.14 Brunner gland hamartoma. This example shows a submucosal-based mass with an overlying intact mucosa composed of nests of benign Brunner glands separated by smooth muscle and vascular septa associated with a focal area of chronic inflammation. |
 FIGURE 3.15 Brunner gland hamartoma. This higher magnification image of Figure 3.14 shows a focus of chronic inflammation, Brunner glands, and associated bands of smooth muscle. |
Finally, and as mentioned at the beginning of the chapter, crushed Brunner glands are a potential pitfall as they can simulate neural/mesenchymal lesions on biopsy. Problematic cases are easily solved with a PAS stain, which will highlight the crushed Brunner glands bright pink (Fig. 3.3, e-Figs. 3.15 and 3.16).
Gastric Hyperplastic Polyps
Occasionally gastric hyperplastic polyps herniate into the proximal duodenum (duodenal bulb) as part of peristalsis, where they can present clinically as duodenal polyps or result in gastric outlet obstruction or even pancreatitis (30). Their appearances are the same as gastric hyperplastic polyps in general (see Volume 2, Chapter 2), consisting of edematous polyps lined by gastric foveolar-type mucosa (e-Figs. 3.57 and 3.58), but such polyps are likely to display prominent features of mucosal prolapse, namely prominent bands of smooth muscle coursing through the polyp (31).
CARCINOMA
Primary Adenocarcinoma
Adenocarcinomas are the most common malignancies of the small intestine (30% to 50% of small bowel malignancies). However, primary adenocarcinomas are still rare lesions, accounting for 2% of GI tract tumors and 1% of GI tract cancer deaths (1). They present in older adults (median age, 67 years), have a male predominance, and are more common in African Americans than in Caucasians.
The vast majority is sporadic and, like sporadic colorectal adenocarcinomas, shares both clinical risk factors and development from adenomatous polyps. The remaining minority of cases arise in the background of certain predisposing conditions, including several of the polyposis syndromes (primarily FAP, MUTYH polyposis, hereditary nonpolyposis colon carcinoma syndrome [HNPCC], PJ syndrome, juvenile polyposis syndrome); Crohn disease; gluten-sensitive enteropathy (GSE) (32); ileostomy; and ileal conduits, among others. Inflammatory bowel disease-associated neoplasia in the small bowel is included in colitis-associated neoplasia in Chapter 4. The greatest risk of small intestinal adenocarcinomas is seen with FAP. In these patients, the relative risk of duodenal adenocarcinoma is striking (relative risk, 330.82) as is that for ampullary adenocarcinoma (relative risk, 123.72) (11). As an aside, there seems to be no significant increased risk for gastric or nonduodenal small intestinal cancer in FAP patients. The risk of small intestinal cancer in Crohn disease and celiac disease are each about 50- to 100-fold.
Both sporadic lesions and predisposing condition-associated lesions are most common in the duodenum, where 65% are periampullary, and their prevalence decreases progressively through the rest of the small intestine. An important exception to the proximal location occurs in patients with Crohn disease, in which 70% of adenocarcinomas are ileal, corresponding to the primary site of the inflammatory process in this disease.
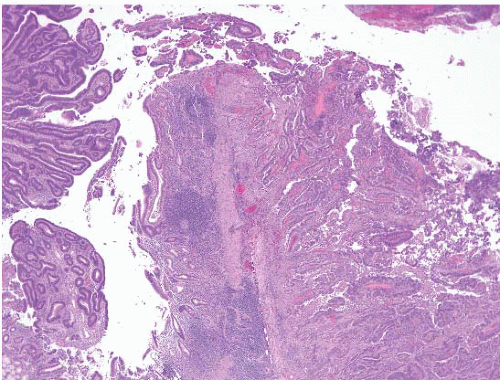 FIGURE 3.16 Small bowel adenocarcinoma. These are histologically similar to colorectal adenocarcinomas. This particular example arose in association with an adenoma (left-hand side of the field) in a patient with familial adenomatous polyposis (FAP). |
Small bowel adenocarcinomas are similar histologically to colorectal adenocarcinomas (Figs. 3.16 and 3.17, e-Figs. 3.59-3.65). Most adenocarcinomas are moderately differentiated, and one-third are poorly differentiated. Degree of differentiation and special histologic subsets [mucinous (e-Fig. 3.66), adenosquamous, sarcomatoid (e-Figs. 3.67-3.70) etc.] have
little bearing on prognosis and so we mention them only as comments when reporting such cases. The majority of small bowel adenocarcinomas have invaded through the bowel wall by the time of diagnosis.
little bearing on prognosis and so we mention them only as comments when reporting such cases. The majority of small bowel adenocarcinomas have invaded through the bowel wall by the time of diagnosis.
 FIGURE 3.17 Small bowel adenocarcinoma. This patient had a history of Crohn disease. Epithelial dysplasia is appreciated on the left-hand side of the field. |
Residual adenomatous epithelium is found with the majority of resected proximal tumors (those likely to be biopsied prior to resection), but often cannot be demonstrated with large distal small intestinal adenocarcinomas, presumably due to tumor overgrowth. Adenomatous epithelium is readily mimicked by tumors metastatic to the GI mucosa and thus the pathologist should be leery of reporting an in situ component, consistently suggesting correlation with appropriate imaging and other studies (2). To this end, immunohistochemistry is used primarily to exclude metastatic disease, specifically metastatic adenocarcinomas [e.g., colon, breast (e-Fig. 3.71), lung] or spread from pancreas cancer or other mimickers of poorly differentiated carcinoma [e.g., melanoma (e-Figs. 3.72-3.78) and lymphoma]. Coordinate labeling for cytokeratins 7 and 20 (CK7 and CK20, Table 3.1) might be helpful in distinguishing small bowel adenocarcinomas from metastatic colon adenocarcinomas. However, such labeling would not distinguish adenocarcinomas from other adjacent sites (such as pancreatobiliary, stomach, lung, ovarian, and endometrial tumors), which may share the same CK7 or CK20 pattern as seen in small bowel adenocarcinomas. Immunohistochemistry should be tailored to differential diagnostic considerations and can frequently help in cases of poorly differentiated malignancies. We have found the use of DPC4 (smad4) antibodies helpful in identifying some cases of pancreatic carcinomas because about 60% of pancreatic carcinomas show loss of this marker in their nuclei (33), a relatively uncommon occurrence in colorectal and small intestinal adenocarcinomas.
Based on SEER data, the overall survival for patients with small bowel adenocarcinoma is about 30% (15).
TABLE 3.1 Cytokeratin 7 and Cytokeratin 20 Expression in Small Intestinal and Colorectal Adenocarcinomas (Ampulla Excluded) | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
Ampullary Carcinomas
The WHO defines ampullary adenocarcinoma as a gland-forming neoplasm usually with either intestinal or pancreatobiliary differentiation, which originates in the ampulla of Vater (15). Essentially, carcinomas from the distal common bile duct, head of the pancreas, or duodenum can all involve the ampulla of Vater, but the term “ampullary carcinoma” should be restricted to carcinomas centered at the ampulla.
Unfortunately, based on the convergence of several structures at the ampulla, ampullary carcinomas are a morphologically heterogeneous group of tumors. Such tumors are rare, accounting for <1% of GI malignant neoplasms (they are rarer than pancreatobiliary neoplasms), in part because the area in which they can arise is small. They are more common in men than women and most patients are in the 7th to 9th decade. Those examples arising in the syndromic setting (FAP, neurofibromatosis) arise in younger patients. Patients present with jaundice, abdominal pain, pancreatitis, and weight loss.
The majority of ampullary carcinomas have an intestinal phenotype, whereas most of the remainder have a pancreatobiliary phenotype (Figs. 3.18 and 3.19, e-Figs. 3.79 and 3.80) (15). A subset has a papillary configuration and rare clear cell, hepatoid, adenosquamous, squamous, undifferentiated, signet ring cell, and undifferentiated with osteoclast-like cell carcinomas have all been reported. For the purposes of diagnosis, it is not so critical to determine the phenotype and subtype as it is to avoid certain pitfalls and to consider metastases or direct spread from adjoining structures. The key step in diagnosis is to assess for features of invasion.
Since intestinal type carcinomas appear similar to colorectal carcinoma, most pathologists are relatively comfortable recognizing them, but once recognized they must be distinguished from metastases from the colorectum (often on clinical grounds). Pancreatobiliary-type lesions can be recognized by searching for desmoplasia, incomplete lumina of neoplastic glands, the presence in a single gland of nuclei with a fourfold difference in size, and luminal necrosis in glands. It is also always worth searching in lacteals and other vascular spaces for tumor cells on mucosal biopsies. As for small bowel and ampullary adenomas, the most critical rule to remember is not to diagnose ampullary carcinomas if there is any degree of uncertainty. It is not difficult for our endoscopy colleagues to obtain additional biopsies from this site, but the consequences of overdiagnosis are severe— such lesions often require a Whipple procedure to adequately resect them. Additionally, it is impossible to know if a carcinoma involving the ampulla on a mucosal biopsy is an “ampullary carcinoma” since tumors restricted to the ampulla have identical appearances and the same variations as lesions of the small intestine and pancreatobiliary tree. The pathologist evaluating an ampullary biopsy showing an adenocarcinoma needs to produce a minimalist report simply stating that there is “adenocarcinoma involving ampullary mucosa”; imaging is required to know where the lesion is centered. Additionally, metastases from a host of other sites have identical appearances and an alarming capacity to mimic an in situ component when they “colonize” the ampullary surface (2) and we are thus extremely hesitant to report ampullary in situ lesions without a note expressing
the capacity of other lesions to mimic in situ neoplasia. We also commonly encounter pancreatic carcinoma with direct spread to the ampulla (Figs. 3.20 and 3.21). Additionally, there exists a subset of intra-ampullary papillary-tubular neoplasm that is analogous to intraductal papillary
neoplasms of the pancreas, but these lesions are typically diagnosed on resection as the entire area must be submitted for histologic evaluation (34).
Since intestinal type carcinomas appear similar to colorectal carcinoma, most pathologists are relatively comfortable recognizing them, but once recognized they must be distinguished from metastases from the colorectum (often on clinical grounds). Pancreatobiliary-type lesions can be recognized by searching for desmoplasia, incomplete lumina of neoplastic glands, the presence in a single gland of nuclei with a fourfold difference in size, and luminal necrosis in glands. It is also always worth searching in lacteals and other vascular spaces for tumor cells on mucosal biopsies. As for small bowel and ampullary adenomas, the most critical rule to remember is not to diagnose ampullary carcinomas if there is any degree of uncertainty. It is not difficult for our endoscopy colleagues to obtain additional biopsies from this site, but the consequences of overdiagnosis are severe— such lesions often require a Whipple procedure to adequately resect them. Additionally, it is impossible to know if a carcinoma involving the ampulla on a mucosal biopsy is an “ampullary carcinoma” since tumors restricted to the ampulla have identical appearances and the same variations as lesions of the small intestine and pancreatobiliary tree. The pathologist evaluating an ampullary biopsy showing an adenocarcinoma needs to produce a minimalist report simply stating that there is “adenocarcinoma involving ampullary mucosa”; imaging is required to know where the lesion is centered. Additionally, metastases from a host of other sites have identical appearances and an alarming capacity to mimic an in situ component when they “colonize” the ampullary surface (2) and we are thus extremely hesitant to report ampullary in situ lesions without a note expressing
the capacity of other lesions to mimic in situ neoplasia. We also commonly encounter pancreatic carcinoma with direct spread to the ampulla (Figs. 3.20 and 3.21). Additionally, there exists a subset of intra-ampullary papillary-tubular neoplasm that is analogous to intraductal papillary
neoplasms of the pancreas, but these lesions are typically diagnosed on resection as the entire area must be submitted for histologic evaluation (34).
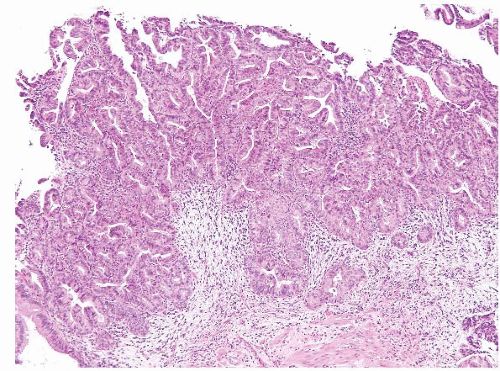 FIGURE 3.18 Ampullary carcinoma. The possibility of metastatic disease or direct spread from adjoining structures should be considered in neoplasms involving the ampulla. |
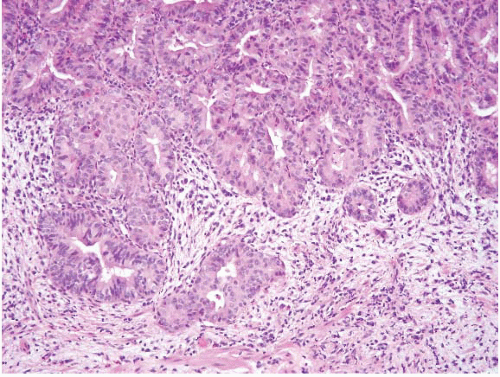 FIGURE 3.19 Ampullary carcinoma. This is a higher magnification image of Figure 3.18. The lesional cells lack intestinal features (no goblet cells) and show no foveolar type mucin. Such tumors have a CK7+/CK20- profile. |
 FIGURE 3.20 Pancreatic carcinoma with direct spread to the ampulla (see Fig. 3.21). There is a normal cluster of ampullary glands on the right of the field. Note rather subtle changes in the neoplastic glands. |
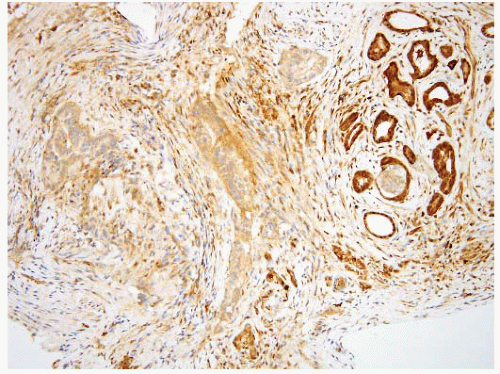 FIGURE 3.21 Pancreatic carcinoma with direct spread to the ampulla. Same case as shown in Figure 3.20, DPC4 immunostain. Loss of nuclear immunolabeling in the carcinoma cells (with retention in nuclei of the ampullary glands at the right of the field) supports the diagnosis of pancreatobiliary adenocarcinoma. An immunoreactive result does not exclude the diagnosis as loss of expression is seen in approximately half of the cases. |
Most ampullary intestinal-type adenocarcinomas express CK20 and about half coexpress CK7, whereas the pancreatobiliary type is CK7+ and CK20-. CDX2 and MUC2 label intestinal-type carcinomas but not pancreatobiliary-type carcinomas. Both types express CEA and CA 19-9 (15). A small percentage is purely mucinous. As repeatedly stressed above, the pathologist must always consider the possibility of metastases from other sites when reviewing such cases.
Metastatic Carcinoma and Other Metastases
As stressed above, the main differential diagnostic consideration of adenocarcinoma in the small bowel is metastatic disease, as the small intestine is the most common GI site for metastases. On purely autopsy-based studies, they outnumber primary carcinomas by over twofold (35) and the small bowel is the favored metastatic site in the GI tract (15). Features that favor a metastatic tumor include the presence of multiple lesions, the absence of a precursor adenoma, a histologic appearance of tumor being “bottom heavy” or encroaching from below, and lack of ulceration. Immunohistochemical studies are also helpful in most circumstances. Common metastatic lesions include melanoma (e-Figs. 3.8 and 3.9), colonic adenocarcinoma (e-Figs. 3.81-3.84), renal cell carcinoma (e-Figs. 3.85-3.87), testicular/germ cell neoplasms (Fig. 3.22, e-Figs. 3.88-3.91), and breast and gynecologic malignancies. Endometriosis is relatively common in the colon, but also can be seen in the small intestine and it mimics metastatic adenocarcinoma. When a malignant neoplasm is encountered in the small bowel of a young male patient, an important consideration is metastatic germ cell tumor from both testis and the retroperitoneum.
An unusual finding in patients who have had intestinal neobladders created to manage urothelial carcinoma is the presence of in situ and invasive urothelial carcinoma in these structures (e-Figs. 3.92-3.95).
NEUROENDOCRINE TUMORS
WDNTs, carcinoid tumors of the small intestine account for about onethird of small intestine neoplasms. They seem to be overrepresented in African Americans (36). Small intestinal WDNTs derive from two separate embryonic divisions of the alimentary tract, the foregut (duodenum) and the midgut (jejunum and ileum). These different origins correlate with distinct neuroendocrine differentiation and with the clinical behavior of these tumors.
Duodenal WDNTs are the second most common tumor in that site; the first is adenocarcinomas. The former represents approximately 20% of GI tract WDNTs (37). Overall, duodenal WDNTs are more common in men (the male-to-female ratio is 1.5:1), with a median age of about 60 years. Two-thirds of duodenal WDNTs are gastrin-producing or G-cell
tumors; the remaining one-third are mostly somatostatin-producing or D-cell tumors, with a small percentage of undefined type. Only one-third of gastrin-expressing tumors are functional and cause Zollinger-Ellison Syndrome (ZES) (see e-Figs. 2.146-2.149 in Chapter 2). These duodenal tumors account for half of ZES cases; the remainder are caused by pancreatic endocrine tumors. One-third of somatostatin-producing tumors are associated with von Recklinghausen disease (NF1) (Fig. 3.23, e-Figs. 3.96-3.99). The somatostatinoma syndrome (i.e., diabetes mellitus, diarrhea, and cholelithiasis), associated with pancreatic islet cell tumors, is virtually never seen in its entirety with these intestinal tumors. Only in the setting of functional tumors, may the suffix “-oma” be appended to refer to such neoplasms (e.g. gastrinoma, somatostatinoma). Proximal small intestinal WDNTs almost never produce the carcinoid syndrome. Functional gastrinomas and somatostatinomas present in slightly younger patients than nonfunctional gastrin-producing and somatostatin-producing tumors, and
are slightly more common in females. Nonfunctional gastrin-producing tumors are usually located in the bulb, while their functional counterparts are found equally in all parts of the duodenum. A subset of the former has been associated with gastric Helicobacter pylori infection and longterm use of proton pump inhibitors. In this setting, the tumors are small G-cell tumors (mean 5.4 mm), located in the bulb mucosa and submucosa, demonstrate an insular pattern on histologic exam, and behave indolently. Some cases demonstrate G-cell hyperplasia in the non-neoplastic mucosa (38). Somatostatinomas typically affect the periampullary region (39).
tumors; the remaining one-third are mostly somatostatin-producing or D-cell tumors, with a small percentage of undefined type. Only one-third of gastrin-expressing tumors are functional and cause Zollinger-Ellison Syndrome (ZES) (see e-Figs. 2.146-2.149 in Chapter 2). These duodenal tumors account for half of ZES cases; the remainder are caused by pancreatic endocrine tumors. One-third of somatostatin-producing tumors are associated with von Recklinghausen disease (NF1) (Fig. 3.23, e-Figs. 3.96-3.99). The somatostatinoma syndrome (i.e., diabetes mellitus, diarrhea, and cholelithiasis), associated with pancreatic islet cell tumors, is virtually never seen in its entirety with these intestinal tumors. Only in the setting of functional tumors, may the suffix “-oma” be appended to refer to such neoplasms (e.g. gastrinoma, somatostatinoma). Proximal small intestinal WDNTs almost never produce the carcinoid syndrome. Functional gastrinomas and somatostatinomas present in slightly younger patients than nonfunctional gastrin-producing and somatostatin-producing tumors, and
are slightly more common in females. Nonfunctional gastrin-producing tumors are usually located in the bulb, while their functional counterparts are found equally in all parts of the duodenum. A subset of the former has been associated with gastric Helicobacter pylori infection and longterm use of proton pump inhibitors. In this setting, the tumors are small G-cell tumors (mean 5.4 mm), located in the bulb mucosa and submucosa, demonstrate an insular pattern on histologic exam, and behave indolently. Some cases demonstrate G-cell hyperplasia in the non-neoplastic mucosa (38). Somatostatinomas typically affect the periampullary region (39).
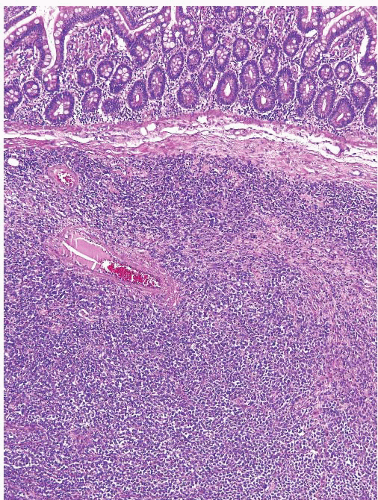 FIGURE 3.22 Metastatic seminoma. Germ cell neoplasms can occasionally involve the small bowel, as seen in this example. Of course, diagnosis requires knowledge of pertinent clinical history and evaluation of immunostains (see e-Figs. 3.110-3.112). |
The few reported cases of high-grade neuroendocrine tumors (small cell carcinomas) have been seen exclusively in men aged about 50 to 80 years; they were typically in the periampullary region and had a dismal prognosis. Brenner et al. retrospectively studied 64 cases of GI tract small cell carcinomas and found that only 3.1% of these arose in the small bowel, the most common primary sites being colon (39%) and esophagus (30%) (40).
Distal small intestinal WDNTs primarily involve the ileum (Figs. 3.24 and 3.25, e-Figs. 3.100 and 3.101), less commonly the jejunum (41), and rarely in association with Meckel diverticula. They account for one fifth of GI tract neuroendocrine tumors and tend to be enterochromaffin cell (EC cell), serotoninproducing WDNTs. The median age of diagnosis is about 60 years and there is no particular sex predilection. The carcinoid syndrome (e.g., watery diarrhea, flushing, and endocardial fibrosis) is seen in 5% to 7% of cases, and jejunoileal WDNTs account for the majority of carcinoid syndrome cases overall. Nearly one-third of patients diagnosed with a small intestinal WDNT have a synchronous or metachronous carcinoma (e.g., colon, stomach, lung, or breast).
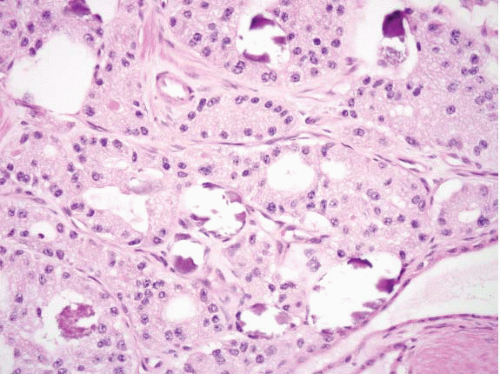 FIGURE 3.23 Somatostatinoma Prominent acinar growth pattern with frequent intraluminal psammoma bodies are seen in this example. |
Duodenal tumors can be readily diagnosed by endoscopy (e-Figs. 3.102-3.108). The location of jejunoileal tumors makes diagnosis more problematic as they often are not amenable to endoscopy/colonoscopy, and conventional imaging techniques (computed tomography and barium contrast studies) are not particularly sensitive. In these circumstances,
radiolabeled somatostatin analogues (octreotide and lanreotide) used in scintigraphy studies have been proven as sensitive detection methods of both primary and metastatic lesions.
radiolabeled somatostatin analogues (octreotide and lanreotide) used in scintigraphy studies have been proven as sensitive detection methods of both primary and metastatic lesions.
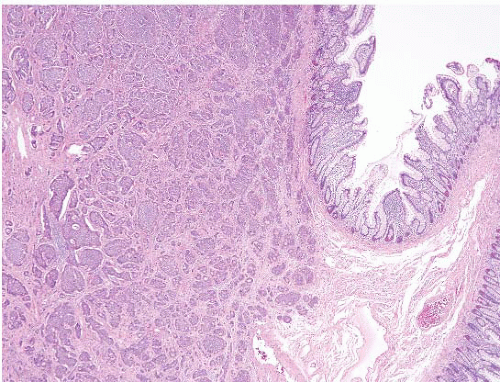 FIGURE 3.24 Ileal well-differentiated neuroendocrine tumor (WDNT). This example shows a prominent nested growth pattern. |
 FIGURE 3.25 Ileal well-differentiated neuroendocrine tumor (WDNT). This higher magnification image of Figure 3.24 shows nests of cells with nuclei displaying salt-and-pepper chromatin pattern and prominent rosette formation. |
Duodenal WDNTs are typically small polypoid lesions (e-Fig. 3.102) that generally measure <2 cm. They are submucosally based lesions with an overlying mucosa that may be focally ulcerated. A focal, subtle mucosal component may be identified on endoscopic biopsy within the lamina propria (e-Figs. 3.1-3.3). Infiltrative larger tumors can occur but are infrequent. G-cell tumors tend to be smaller than D-cell tumors and a minority (15%) are multicentric.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


